Tous les organismes
–excepté les virus – sont constitués de cellules comportant une membrane
extérieure composée de lipides, et un génome,
composé d'acides nucléiques, comprenant l'ensemble des instructions nécessaires
pour fabriquer l’organisme. Ces instructions ont pour support les gènes, disposés de façon linéaire sur
les chromosomes de chaque cellule. La plus simple des bactéries libres contient
par exemple un génome d'environ deux mille gènes, qui suffit à définir
l'organisme tout entier. Chaque gène
occupe une place, ou locus, sur le chromosome. Le matériel génétique est
l'acide désoxyribonucléique, ou ADN, molécule du chromosome.
L'ADN est une chaîne
linéaire composée de quatre types d'unités chimiques (les nucléotides, abrégés
en A, T, C et G) qui peuvent se suivre dans n'importe quel ordre. La structure
de l'ADN est une hélice à deux brins. Un brin contient la séquence d'un gène et
l'autre brin une séquence complémentaire déterminée par les règles
d'appariement des quatre nucléotides (A s'apparie avec T, et C avec G). Chaque
chromosome d'une cellule contient une molécule d'ADN compactée. Chaque gène est
un segment de la molécule d'ADN du
chromosome.
Les gènes agissent par
l'intermédiaire des molécules qu'ils produisent. Les produits directs d'un gène
sont des molécules d'acide ribonucléique (ARN). En effet, lorsqu'un gène est
actif, i.e. lorsqu'il s'«exprime», il est recopié en un brin d'ARN par un
processus appelé transcription, et
cette copie est appelée ARN messager ou ARNm.
Un ARNm est responsable
de la synthèse d'une protéine, ou "traduction",
qui est effectuée par une structure appelée ribosome. Les protéines sont des
chaînes linéaires d'acides aminés, dont
il existe une vingtaine de formes. La séquence de nucléotides de l'ARN
détermine la séquence d'acides aminés de la protéine. La relation entre la
séquence des nucléotides d'un gène et la séquence des acides aminés dans la
protéine correspondante est donnée par le code génétique : chaque acide aminé
est codé par trois nucléotides, appelés triplets, et certains triplets de
nucléotides codent pour le même acide aminé. Le ribosome glisse le long du brin
d'ARN messager pour lire la succession de triplets, et construit la chaîne
d'acides aminés correspondante, jusqu'à synthèse totale de la protéine. La
nouvelle chaîne d'acides aminés quitte ensuite le ribosome et se replie sur
elle-même dans une configuration caractéristique, déterminée par la séquence
des acides aminés. C'est la forme tridimensionnelle de la protéine qui
détermine sa fonction chimique à l'intérieur de l'organisme.
Chaque gène est donc défini
d’une part par sa structure (fragment d’ADN) qui détermine la synthèse d’une
protéine donnée, et par sa localisation sur un chromosome d’autre part.
Mais les nucléotides de
l'ADN qui codent pour la structure des protéines ne sont pas les seuls constituants
des gènes. Il existe en effet des groupes
de nucléotides adjacents aux séquences de codage qui contrôlent la quantité
et le devenir des produits des gènes. Certaines de ces séquences régulent la
transcription du gène et ne sont pas transcrites. D'autres régulent la
traduction et se retrouvent dans l'ARN messager, de part et d'autre de la
séquence codant pour la protéine : on appelle ces séquences, non traduites,
"3' UTR" et
"5' UTR" selon l'extrémité de l'ARNm où elles se trouvent.
De plus, chez les
organismes eucaryotes, une séquence de nucléotides codant pour une protéine
peut être interrompue par des séquences non codantes, appelées introns. Pendant la transcription, les
introns sont reproduits le long de l'ARN avec les séquences codantes, produisant
une molécule d'ARN géante, appelée ARN pré-messager. Les séquences
correspondant aux introns sont ensuite extraites de l'ARN par un processus
appelé excision-épissage. L'ARN pré-messager subit aussi deux autres
modifications :
-
à l'une de ses
extrémités (dite 5'), l'ARN reçoit une "coiffe"
-
à l'autre extrémité
(dite 3'), sa séquence de nucléotides est allongée par une série de A appelée
"queue polyA" (polyadénylation). Cet ajout protégerait l'ARN messager
d'une dégradation trop rapide pendant la traduction.
Ainsi, chez les
eucaryotes, les ARN pré-messagers subissent une maturation avant de diriger la synthèse des protéines.
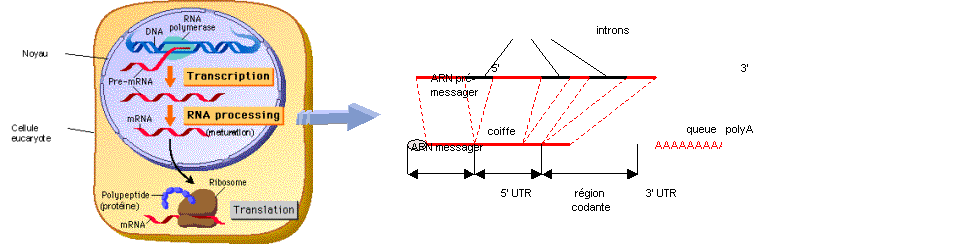
Le schéma suivant (cf. [1]) résume la façon
dont un gène s'exprime dans une cellule eucaryote :
Figure 1
: du gène à la protéine, dans une cellule eucaryote
Les gènes contrôlent
donc la formation des protéines, composés fondamentaux pour tous les processus
biologiques. Les protéines sont non seulement les constituants majeurs de la
plupart des structures cellulaires, mais contrôlent également la quasi-totalité
des réactions chimiques qui ont lieu chez les organismes vivants. La protéine
intervient soit comme élément structural, soit comme enzyme modifiant la
vitesse d'une réaction chimique.
Toutes les cellules d'un
même organisme contiennent les mêmes gènes, mais synthétisent des protéines
différentes. Les différents types de cellules (musculaires, nerveuses, etc.)
doivent donc avoir une combinaison particulière de gènes actifs pendant que
d'autres restent inactifs. Dans un type de cellule donné chez l'Homme,
on estime qu'approximativement 10 à 15 000 gènes sont effectivement exprimés
sur les 30 000 à 60 000 gènes de notre génome. On a de plus des différences de
niveau d'expression au sein de ces gènes effectivement exprimés : d'un point de
vue quantitatif, il faut savoir que certains gènes sont exprimés à quelques
centaines voire quelques milliers d'ARNm par cellule, alors que la majorité des
gènes exprimés n'est présente qu'à un faible nombre d'ARNm par cellule.
La progression d'une
cellule d'un état vers un autre état, pathologique par exemple, correspond
souvent à des changements qualitatifs et quantitatifs dans l'expression des
gènes, et donc dans les populations d'ARNm et de protéines de cette cellule,
comme l'illustre le schéma suivant (cf. [7]) :
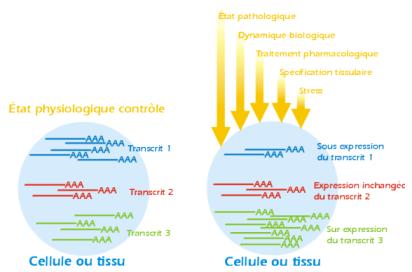
Figure 2
: variations de l'expression des gènes
En particulier, l'état tumoral d'une cellule correspond à
une division cellulaire accélérée : certaines protéines (et donc certains ARNm)
doivent donc être différentiellement exprimés par rapport à une cellule saine
de même type. Identifier et étudier ces protéines représente un enjeu important
pour une meilleure compréhension des phénomènes moléculaires associés aux
cancers. C'est dans cette optique que se situe le projet d'étude de
l'expression des gènes dans les cancers des VADS (Voies Aéro-Digestives
Supérieures), mené à l'IGBMC par l'équipe du Dr. B. Wasylyk, et auquel j'ai
participé sur le plan de la bioinformatique.
Le terme de cancers des VADS regroupe les tumeurs
développées au niveau de la bouche, du pharynx, des fosses nasales, des sinus
et du larynx.
Ce type de cancer représente un problème majeur de santé
publique au niveau mondial : on estime le nombre de nouveaux cas par an
dans le monde à 540 000 (cf. [9]). La fréquence des cancers de la cavité
buccale en Inde et celle des cancers du naso-pharynx en Asie du Sud-Est sont
particulièrement élevées, mais l’Europe reste le continent le plus touché avec
26 nouveaux cas pour 100 000 personnes par an. La France occupe le premier rang
mondial avec 60.8 nouveaux cas pour 100 000 personnes par an. C’est dans le
Nord-Pas-de-Calais et dans le Nord-Est que l’on observe les plus fortes
fréquences : dans le Bas-Rhin en particulier, cette fréquence est de 63.6
nouveaux cas pour 100 000 personnes par an. Les cancers des VADS représentent
en France 12 % des causes de mortalité par cancer chez l’homme, et la survie à
5 ans ne dépasse pas 30 %, avec des fréquences élevées de récidive locale ou
régionale (50 %), ou de métastase à distance (30 %).
Si l’on sait que la progression vers un état tumoral repose
sur l’exposition à des cancérigènes, et est liée à des facteurs irritatifs
locaux (l’alcoolisme chronique et le tabagisme d’une part, et les traumatismes
répétés dans la bouche par des problèmes dentaires d’autre part),
l’amélioration de la prise en charge clinique et thérapeutique de ces cancers
nécessite cependant une meilleure description des événements génétiques
associés à cette progression tumorale.
L’étude menée a pour objectif la caractérisation des
événements moléculaires et l’identification de nouveaux gènes impliqués dans
les cancers des VADS, ce qui pourrait permettre à plus long terme le
développement de nouveaux marqueurs pronostiques et diagnostiques, voire de
nouvelles perspectives thérapeutiques.
Cette étude est menée conjointement par :
-
le laboratoire du Dr J. Abecassis (Centre Paul Strauss,
Strasbourg), qui fournit les échantillons sains et tumoraux et détermine la
fréquence des événements identifiés sur un nombre plus significatifs
d’échantillons
-
la société Exonhit Therapeutics, propriétaire de la
technologie génomique DATAS, qui recherche des profils qualitatifs d’expression
génique, afin d’identifier les événements d’épissage alternatif qui peuvent se
produire dans les cellules au cours de leur progression vers un état tumoral
-
le laboratoire du Dr. B. Wasylyk (IGBMC, Strasbourg), qui
recherche des profils quantitatifs d’expression génique, afin d’identifier les
ARNm (ou « transcrits ») sélectivement surexprimés ou réprimés lors
de l’évolution tumorale des cellules
Les données à traiter en bioinformatique sont celles du
laboratoire du Dr. B. Wasylyk.
La recherche des profils quantitatifs d’expression génique,
menée par l’équipe de Dr. B. Wasylyk, consiste à comparer les niveaux
d’expression des gènes dans des échantillons tumoraux et dans des échantillons
sains.
Le Centre Paul Strauss, membre du Centre National de
Recherche contre le Cancer, a fourni des ARN totaux de cellules prélevées chez
des patients atteints d’un cancer des VADS. Ces cellules ont été classifiées
selon le stade d’évolution tumorale et le comportement clinique :
·
E (early) : les cellules proviennent de tumeurs de
petite taille, peu différenciées, et ont été prélevées par chirurgie à un stade
relativement précoce de développement du cancer.
·
S (stable) : les cellules proviennent de tumeurs
de taille moyenne, et sont différenciées de façon homogène. Après exérèse
chirurgicale de la tumeur, il n’a pas été observé de métastase.
·
U (unstable) : les cellules proviennent également
de tumeurs de taille moyenne, mais sont différenciées de façon hétérogène. De
plus, après exérèse de la tumeur, des seconds cancers ou des extensions
métastasiques ont été observées.
·
N (normal) : ce sont des cellules prélevées sur
des tissus normaux (luette). On distingue dans certains cas les cellules de
type NE (cellules saines prélevées chez un patient atteint d'un cancer des VADS
de type E), NS et NU.
L’intérêt de cette classification est de permettre :
·
d’une manière générale, la caractérisation des gènes
qui s’expriment différentiellement dans des cellules saines et dans des cellules
tumorales,
·
l’identification de gènes impliqués dans les étapes
précoces de l’évolution du cancer,
·
l’identification de marqueurs moléculaires permettant
la distinction entre les tumeurs de bon et de mauvais pronostic, de façon à
pouvoir adapter le protocole thérapeutique après une biopsie.
La détermination des profils d’expression des gènes dans les
types de cellules décrits s’appuie sur différentes techniques.
1.4.2.1
Puces à ADN Affymetrix™
Le principe de cette
technique repose sur l'utilisation de réseaux d'ADN (membranes haute-densité ou
puces à ADN) constituées de plusieurs milliers de sites appelés « unités
d'hybridation ». Chaque unité d'hybridation est composée de plusieurs
millions d'exemplaires d'une même molécule d'ADN dénommée « cible »,
immobilisée sur le support solide de la puce.
La première étape pour
quantifier l’expression des gènes d’une cellule consiste à obtenir des cDNA par
transcription inverse des ARNm extraits de cette cellule. Ces cDNA sont ensuite
marqués par fluorescence ou par radioactivité, et sont appelés «sondes
complexes». S'il y a complémentarité entre une cible donnée et un cDNA marqué,
il y aura formation d'un duplexe cDNA marqué/cible, ou hybridation ou
appariement. Un système de lecture adéquat permet ensuite d'identifier et de
quantifier sur chaque unité d'hybridation le signal émis (radioactivité ou
fluorescence). L'intensité du signal d'hybridation mesuré reflète l'abondance
du transcrit correspondant dans la population d'ARNm qui a servi à préparer la
sonde complexe. Ainsi, une cible émettant un fort signal d'hybridation
correspond à un gène fortement exprimé. Une puce à ADN permet donc d'étudier le
profil d'expression de plusieurs milliers de gènes à la fois (de tous les gènes
représentés sur la puce par des cibles). Le schéma suivant résume ce principe
(cf. [7]) :
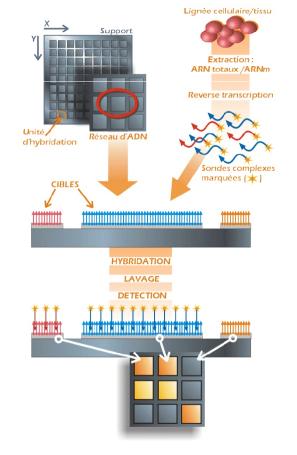
Figure 3
: principe des puces à ADN
Ici, pour chaque échantillon de cellules, on réalise cette
expérience avec une puce à ADN commercialisée par la société Affymetrix™
permettant d’identifier 12 650 ARNm – donc 12650 gènes – différents (avec 32
unités d’hybridation par ARNm). Les puces utilisées pour les différents
échantillons sont identiques. Ainsi, pour chaque ARNm identifiable, on obtient
un profil d'expression, c’est-à-dire ici une suite de chiffres reflétant les
quantités de cet ARNm détectées par les puces, donc les quantités présentes
dans les différents échantillons.
1.4.2.2
Differential Display
Cette technique est fondée sur la séparation sur gel des parties 3' terminales des ARN messagers
extraits de cellules, ou RT-PCR (Reverse Transcription Polymerase Chain
Reaction). Comme l'indique le schéma ci-dessous (cf. [7]), la première étape
consiste à synthétiser les cDNA complémentaires des parties 3' terminales des ARNm
par la réaction de transcription inverse : l'amorce utilisée, un
oligonucléotide polydT (…TTTTTT), est spécifique de la queue polyA située à
l'extrémité 3' des ARNm. Les cDNA ainsi obtenus sont amplifiés par PCR, en
utilisant différents couples d'amorces constitués d'une part de l'amorce polydT
précédemment utilisée, et d'autre part d'une amorce arbitraire. On obtient des
produits de PCR différents selon le couple d'amorces utilisé : pour une amorce
arbitraire donnée, seuls les cDNA contenant une partie de séquence
complémentaire de cette amorce seront amplifiés. Pour chaque couple d'amorces,
on sépare les produits de PCR sur un gel d'électrophorèse : les cDNA amplifiés
migrent sur le gel en fonction de leur taille. On obtient ainsi des bandes à
différents endroits sur le gel : chaque bande contient un ou plusieurs cDNA de
même taille.
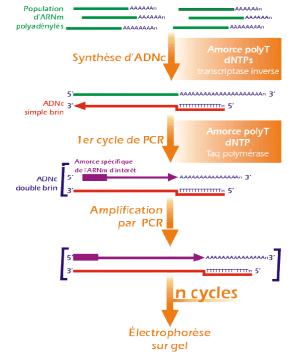
Figure 4
: principe de la RT-PCR
Ceci est réalisé en parallèle pour 2 échantillons de chaque
type (NE, E, NU, U, NS, S). Ainsi, pour chaque couples d'amorces, donc sur
chaque gel d'électrophorèse, on fait migrer en même temps les produits des 12
PCR menées parallèlement. On obtient de cette façon des gels ayant l'allure
suivante :

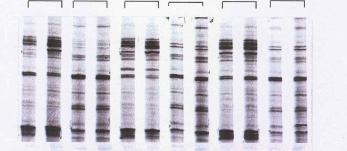
Figure 5
: exemple de gel de Differential Display
Le terme "Differential Display" vient du fait
qu'avec cette méthode, on peut comparer les quantités d'un ARNm donné présentes
dans les différents types de cellules. Comme sur la figure 5 par exemple, si
l'on observe une bande dans la colonne des cDNA issus de cellules de type U,
alors que cette bande est peu visible dans la colonne des cDNA issus de
cellules de type NU, on pourra supposer que l'ARNm (ou les ARNm) correspondant
à cette bande est beaucoup plus présent dans les cellules tumorales d'un cancer
de type instable, que dans les cellules saines.
On connaît ainsi le profil d'expression de chaque bande (ou
"ligne"), c'est-à-dire ici une suite de 6 chiffres correspondant aux
6 intensités de la bande dans les 6 paires de colonnes du gel. Cette technique
permet donc d'obtenir le profil d'expression de la majorité des ARNm présents
dans les cellules (95 % des ARNm en théorie si les amorces sont bien choisies).
Cette technique permet de fixer sur une membrane et
d'étudier la spécificité d'expression (cellules saines ou cancéreuses) de
plusieurs ARNm en même temps.
Les cDNA correspondant aux différents ARNm à étudier (ici le
ou les cDNA issus d'une même bande de Differential Display) sont clonés puis introduits
sous forme de plasmides dans des bactéries d'espèce E. coli. Ces bactéries sont cultivées et forment des colonies,
chaque colonie contenant spécifiquement l'un des cDNA (ou clones) étudiés. Un
certain nombre de colonies (ici 4 ou 8 selon l'expérimentateur) sont prélevées
et placées dans une plaque. Le nombre de colonies prélevées doit être
suffisamment grand pour que les clones ou cDNA à étudier soient tous
représentés – pas nécessairement de façon unique – dans ces colonies
prélevées. On génère ensuite une copie conforme de la plaque sur une membrane,
où les colonies subissent une série de traitements de sorte à fixer les
plasmides qu'elles contiennent (dénaturation, neutralisation, lavage, séchage,
fixation). La membrane est ensuite incubée avec une sonde radioactive.
Ici, on réalise deux fois cette expérience pour chaque bande
étudiée ; l'une des deux membranes est incubée avec une sonde dite
"normale", l'autre avec une sonde dite "tumorale". La sonde
tumorale est générée à partir des échantillons tumoraux de type E : les
ARNm de ce type de cellules sont extraits, puis soumis à la réaction de
transcription inverse. Les cDNA obtenus sont amplifiés et marqués
radioactivement. La sonde tumorale est donc constituée d’un ensemble de cDNA
radioactifs correspondant aux ARNm d’une cellule tumorale. La sonde normale est
générée de la même façon à partir d’échantillons de type NE.
Ainsi, si un clone est hybridé en Reverse Northern avec la
sonde tumorale, mais beaucoup moins avec la sonde normale, alors cela signifie
que la séquence complémentaire de ce clone était beaucoup plus représentée dans
les cDNA de la sonde tumorale. Un tel clone correspond donc vraisemblablement à
un ARNm surexprimé dans les cellules cancéreuses. On obtient par exemple un
résultat de ce type pour deux des 8 clones issus de la bande notée A 0452 :





Figure 6
: exemple de résultats de Reverse Northern
1.4.2.4
Northern blot
Cette technique consiste à extraire les ARNm des cellules à
étudier, puis à les séparer en fonction de leur taille sur gel
d'électrophorèse, puis à les transférer sur une membrane (filtre en
nitrocellulose ou nylon) de sorte à obtenir une empreinte du gel sur la membrane. L'identification et la
quantification d'un ARNm donné sont réalisées par appariement de cet ARNm avec une sonde d'ADN, c'est-à-dire ici
un court fragment d'ADN dont la séquence est complémentaire de celle de l’ARNm
à étudier.
Durant la première étape (électrophorèse), les différents
ARNm vont migrer à des vitesses différentes selon leur taille. Après migration,
les ARN pourront être visualisés grâce par fluorescence après exposition aux
ultraviolets. On peut ainsi observer pour chaque échantillon une fluorescence
diffuse sur toute la longueur de la piste de migration, témoignant de la
présence d'ARNm de différentes tailles. Le gel est ensuite placé dans une
solution tampon et couvert d'un filtre de nitrocellulose (ou nylon) et d'une
pile de serviettes en papier. Les fragments d'ARN sont transférés vers le
filtre par le tampon qui est absorbé par les serviettes en papier (transfert
par capillarité, pendant 16 heures). Le filtre est enlevé et incubé avec une
sonde d'ADN monobrin marquée radioactivement, et complémentaire de la séquence
de l'ARNm recherché. Après appariement (hybridation), la sonde non liée est
éliminée par lavages, et le filtre est exposé à un film sensible aux rayons X
(autoradiographie) émis par la sonde. Comme la sonde ne s'est appariée qu'aux
fragments d'ARN dont elle est complémentaire, le film ne sera impressionné que
par les bandes qui y correspondent. On obtient ainsi le profil d'expression de
l'ARNm sondé, c'est-à-dire la suite des intensités des bandes détectées pour
les différents échantillons.
Le schéma suivant résume cette technique (cf. [7]) :
On
visualise les trois bandes correspondant à l'ARNm sondé. Leurs intensités
reflètent les quantités de cet ARNm présente dans les différents échantillons
de cellules.
|
|
Sur le
gel d'électrophorèse, les ARNm issus d'un même échantillon de cellules se
trouvent dans la même colonne de migration.
|
|
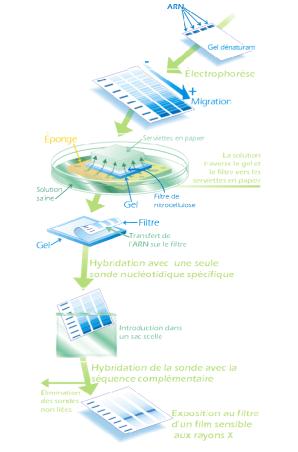
Figure 7
: principe du Northern Blot
Cette technique permet ainsi d'obtenir le profil
d'expression d'un seul ARNm à la fois, contrairement aux techniques précédentes.
1.4.2.5
Virtual Northern
Le principe de cette technique est sensiblement le même que
pour la technique précédente. La différence réside dans le fait que les ARNm
extraits des cellules sont préalablement soumis à la réaction de transcription
inverse, et que les cDNA obtenus sont amplifiés. Ce sont ces cDNA amplifiés que
l’on fait migrer sur un gel d’électrophorèse. Ceci permet de réduire
considérablement la quantité d’ARNm nécessaire.
Deux séries d'expériences sont menées indépendamment :
·
la série
d'expériences "Affymetrix" :
-
La première expérience menée en utilisant les puces à ADN
Affymetrix a été réalisée à partir de 10 échantillons (10 patients) : parmi
ceux-ci, 2 sont de type normal (N), 2 de type précoce (E), 3 de type stable (S)
et 3 de type instable (U). On a donc, pour chacun des 12 650 ARNm détectables,
10 chiffres représentant les niveaux d’expression de cet ARNm dans les
différents échantillons ou chez les différents patients. Ces données sont tout
d’abord normalisées, puis analysées par ANOVA (méthode statistique d’analyse de
la variance) en considérant le type d’échantillon comme facteur. Cela permet de
détecter les gènes dont l’expression varie significativement entre les
différents types d’échantillons. On sélectionne ainsi 1 600 ARNm environ. Un
traitement de « clustering » est ensuite réalisé sur ces 1600
ARNm : on regroupe les ARNm présentant des profils d'expression
similaires. On obtient alors 14 groupes, chaque groupe étant caractérisé par un
profil-type.
-
Une seconde expérience similaire a été réalisée par la suite,
avec 10 nouveaux échantillons. Le traitement statistique des profils obtenus
n'est pas encore terminé.
·
la série
d'expériences "Differential Display – Northerns" :
Les besoins exprimés par l'équipe du Dr. B. Wasylyk sont :
·
de regrouper, de comparer et d'exploiter :
-
les résultats de toutes les expériences d'analyse d'expression
génique, c'est-à-dire les profils d'expression des gènes des puces Affymetrix,
des bandes de Differential Display, des clones traités en Northerns
-
les résultats d'analyse manuelle et automatique des séquences
des clones.
·
de déterminer automatiquement la localisation chromosomique
et la fonction des gènes correspondant aux clones étudiés.
Or les résultats des expériences précédemment décrites ont
jusqu'à présent été stockées sous diverses formes :
-
les profils d'expression des gènes des puces Affymetrix
sélectionnés par ANOVA sont consignés dans un fichier texte
-
les résultats de Differential Display, Northern Blot, Reverse
Northern, Virtual Northern, ainsi que les résultats d'analyse manuelle de
séquences, sont stockés de façon différente selon l'expérimentateur : ces
données se répartissent en 4 fichiers Access, 2 fichiers Excel et 1 fichier
Word. Au sein de ces fichiers, on retrouve des champs ou colonnes communes
("user", numéro de clone, résultats de Differential Display, …). Mais
au sein de ces colonnes, les types de données varient (par exemple, les résultats de Differential Display se présentent
sous forme de chiffres ou bien de lettres). Il existe de plus des champs
spécifiques à chaque expérimentateur : c'est notamment le cas pour les
résultats de Reverse et Virtual Northern.
Cette diversité de formats ne permet pas d'exploiter
globalement les résultats obtenus. Un outil informatique intégrant l’analyse
automatique de séquences et la gestion de toutes les données concernant le
projet – y compris les résultats de cette analyse de séquences – s’avère donc
nécessaire.
Cet outil doit en
premier lieu être utilisé par l'équipe du Dr. B. Wasylyk, et à plus long terme
également par les autres partenaires du projet. Il devra aussi préserver la
confidentialité des données, en limitant l'accès aux données aux utilisateurs
autorisés, et en limitant leurs droits.
Un outil informatique dédié à l’analyse de génomes et de
séquences protéiques (G-scope) ayant déjà été développé par le laboratoire de
Génomique Structurale, la solution retenue pour répondre aux besoins décrits
est d’utiliser cet outil et de l’adapter à l’analyse de séquences nucléiques,
la gestion des données étant assurée par une base de données relationnelle. Ces
deux « modules » sont bien sûr interdépendants : la base
de données contient les séquences à analyser pour G-scope, et doit également
recevoir les résultats de cette analyse.
La solution retenue est le système de gestion de bases de
données relationnelles (SGBD-R) Microsoft® Access 2000. En effet, la gestion
des données via un SGBD (plutôt que sous forme de système de fichiers) permet :
-
une mise à jour et une interrogation aisée : l'utilisateur ne
se préoccupe pas de l'implantation physique des données.
-
la gestion de la cohérence et de l'intégrité des données : des
critères précis, ou contraintes d'intégrité, concernant la validité des données
peuvent être décrits par l'utilisateur. Ces critères sont systématiquement
contrôlés par le SGBD.
-
la non-redondance : une information n'est stockée qu'une seule
fois, ou, exceptionnellement, avec une redondance calculée, ce qui facilite la
mise à jour et le maintien de la cohérence des données
-
la gestion du partage des données dans un environnement
multi-utilisateurs (gestion des accès simultanés).
-
la gestion de la sécurité : protection contre les accès non
autorisés, personnalisation des droits d'accès par groupe d'utilisateurs, ou
même par utilisateur.
Le choix d'un modèle relationnel permet :
-
l'utilisation de structures de données simples pour stocker
les différents types d'entités manipulées : les tables,
-
une organisation reflétant les liens entre les entités, grâce
aux liens établis entre les tables,
-
pour la gestion de la structure de la base et pour la
manipulation des données, l'utilisation d'un langage (SQL) standard de
haut niveau, c'est-à-dire non
procédural : on ne précise pas l'algorithme d'accès aux données, mais on donne
seulement les critères de sélection des données recherchées.
Trois entités se dégagent de l'analyse des besoins :
-
le gène des puces Affymetrix,
-
la bande de Differential Display,
-
le clone traité en Northern blot, Reverse et Virtual Northern,
et dont la séquence est à analyser.
Ces entités sont associées de la façon suivante :
-
un clone provient d'une et une seule bande, et une bande donne
un ou plusieurs clones. On dit que cette association est de type (1,n), puisque
pour un clone donné, on n’a au maximum qu’une bande, alors que pour une bande
donnée, on a une liste de n clones.
-
un clone correspond à 0 ou 1 ou n gènes des puces Affymetrix ;
et réciproquement, un gène correspond à 0 ou n clones ("correspond"
signifie ici "est localisé au même endroit sur le génome humain"). On
dit que cette association est de type (n,n), puisque pour un clone donné, on a
une liste de gènes Affymetrix, et réciproquement.
On a donc le schéma conceptuel Entité-Association suivant :
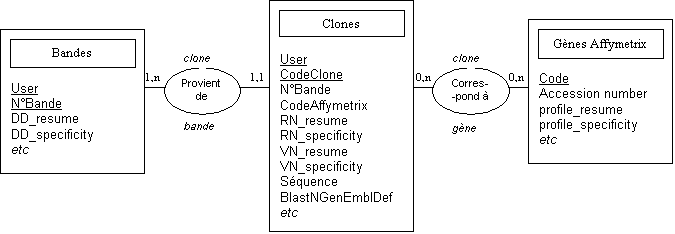
Figure 8
: schéma conceptuel Entités –Associations
Le schéma logique relationnel correspondant à ce schéma
Entités-Associations (et utilisé dans Access) est plus complexe. Du fait tout
d’abord de l’association « Correspond à » de type (n,n), il faut
ajouter une table (nommée « Overlaps ») permettant de stocker
plusieurs codes de gènes Affymetrix pour un même clone. Il est en effet
impossible de stocker une liste (de codes de gènes Affymetrix ici) dans un
champ d’une base de données relationnelle. La solution consiste donc à
introduire une table supplémentaire, où les listes de n codes Affymetrix sont
stockées sous forme de n lignes (ou enregistrements), chaque ligne étant
constituée d’un code de clone et d’un code de gène Affymetrix.
De plus, il existe pour chaque clone des champs communs et
des champs spécifiques à l'expérimentateur. En effet, regrouper l'ensemble de
ces champs dans la table Clones aurait conduit à une table contenant une
centaine de champs, dont seulement un tiers
aurait été renseigné pour chaque ligne. La table Clones ne contient donc
que les champs communs à tous les expérimentateurs, et une table supplémentaire
par expérimentateur est créée, contenant ses champs spécifiques.
On a donc le schéma relationnel suivant :
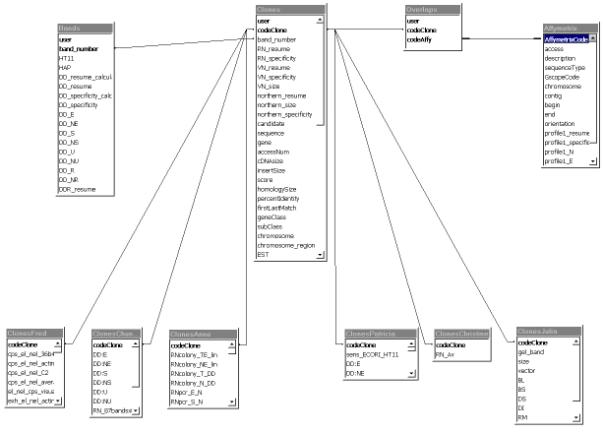
Figure 9
: schéma logique relationnel
Les champs en gras constituent les clés primaires de chacune
des tables, c’est-à-dire les informations identifiant chaque ligne de façon
unique.
Remarque : pour les tables Clones et Bandes, il est
nécessaire d'utiliser une clé primaire composée de deux champs : le code
attribué par l'expérimentateur, et nom de l'expérimentateur. En effet, chaque
expérimentateur a numéroté ses bandes et ses clones indépendamment des autres,
et il est donc possible d'avoir plusieurs clones ou bandes avec le même code.
Le code VBA associé à la base de données comporte
différents types de procédures :
- Procédure
liée au regroupement et au
formatage des résultats :
-
transfert des données depuis les fichiers personnels des
expérimentateurs vers la base de données commune
- Procédures
liées à l’analyse des profils
d’expression :
-
calcul des résumés de profils d’expression : pour chaque
bande de Differential Display et pour chaque gène des puces Affymetrix, on
calcule une valeur représentant son profil d’expression, à partir de ses
niveaux d’expression dans les différents types de cellules (N, E, S, U). Cette
valeur spécifie si la bande ou le gène s’exprime spécifiquement dans les
cellules saines (profil sain, noté « N ») ou dans les cellules
tumorales (profil tumoral, noté « T »), ou bien s’il impossible de
dégager une spécificité T/N (profil « C », complexe).
-
calcul de la spécificité vis-à-vis d’un ou deux types de
cancer pour les bandes ou gènes de profil T ou C. Cette spécificité peut être
E, S, U, E_S, S_E, E_U, U_E, U_S, ou encore S_U: il s'agit du ou des deux types
de cancer dans lequel la bande ou le gène Affymetrix s'exprime le plus.
- Procédures
liées à l’automatisation de
l’analyse de séquences :
-
transfert des séquences des clones, sous forme de fichiers au
format FastA, vers le répertoire de travail de G-scope
-
transfert des numéros d’accession dans GenEmbl des gènes des puces Affymetrix sélectionnés par
l’ANOVA, vers le répertoire de travail de G-scope
-
transfert des
résultats de l’analyse automatique de séquences réalisée avec G-scope vers la
base de données.
- Procédure
liée à la validation manuelle de
l’analyse de séquences :
-
visualisation des fichiers générés sous G-scope
(exemple : fichier de BlastN contre le génome humain)
Une interface homme-machine est associée à ces
procédures : elles peuvent être exécutées facilement depuis l’application
Access.
L’analyse de séquences s’appuie essentiellement sur la
recherche de séquences similaires dans des banques de séquences.
C’est le développement récent des techniques de séquençage
de l'ADN qui a permis la création de ces énormes banques de séquences. A ce
jour, les banques de données publiques contiennent par exemple les génomes
complets de plus de 30 espèces : ainsi, les génomes de 22 bactéries, 7 archae,
1 eucaryote inférieur (la levure), et de certains eucaryotes supérieurs (comme
le nématode C. elegans ou la plante Arabidopsis Thaliana) ont été
entièrement séquencés. Le génome de l'homme est en majeure partie disponible.
De plus en plus de génomes complets sont régulièrement publiés (environ un par
mois).
Il est possible de détecter et de prédire les protéines
codées dans ces génomes. Si la fonction n'est connue que pour quelques dizaines
de milliers de ces protéines, près de la moitié des fonctions peuvent être
inférées par analyse de séquence (recherche de séquences homologues dans les
banques). Dans le cas particulier du génome humain, on estime que les 3.3
milliards de bases de ce génome devraient contenir 30 000 à 60 000 gènes et
donc quasiment autant de protéines (et seules 500 d'entre elles seraient les
cibles de l'ensemble des médicaments actuellement sur le marché…).
Il existe deux grands types de banques de données (cf. [8])
:
·
les banques de
séquences nucléiques : outre les génomes complets, précédemment cités, on
trouve dans cette catégorie GenBank
et EMBL (regroupées localement à l'IGBMC sous le nom de "GenEmbl"),
ainsi que les ESTs. La banque des ESTs (Expressed Sequence Tags) contient des
fragments de cDNA obtenus lors d’études d’expression des gènes. C’est donc la
banque qui présente le plus de « réalité biologique », puisqu’elle ne
contient pas de séquences prédites, mais seulement des séquences réellement
observées. EMBL est la première base de séquences nucléiques en Europe ; elle
est contrôlée et distribuée sur CD-Rom ou via Internet
par l'Institut Européen de Bioinformatique (EBI). GenBank est l'équivalent
américain de la base EMBL ; ses séquences sont fournies par des laboratoires ou
proviennent de projets de séquençage à grande échelle.
A ce jour, la banque "GenEmbl" contient
environ 4 millions de séquences, et celle des ESTs en contient plus de 8
millions.
·
les banques de
séquences protéiques : les principales banques de cette catégorie sont
SwissProt, SpTrEMBL et la banque de structures spatiales PDB (regroupées
localement sous le nom de "Protein"). Les protéines contenues dans
SpTrEMBL sont les traductions automatiques des séquences codantes extraites de
la banque nucléique EMBL. De telles séquences peuvent ensuite être incorporées
dans SwissProt après avoir été vérifiées et annotées.
Ainsi, la banque "Protein" contient aussi
bien des séquences de protéines étudiées biologiquement que celles de protéines
prédites à partir de séquences nucléiques. Elle contient plus de 690 000
séquences à ce jour.
Ces différentes banques se recouvrent les unes les autres,
ne sont pas non redondantes, et contiennent encore beaucoup d'erreurs (de
séquences et d'annotation).
Le protocole
d’analyse retenu fait intervenir différents programmes, développés à l’IGBMC ou
disponibles dans le domaine public. Parmi ces programmes, le « chef
d’orchestre » est G-scope.
2.2.2.1 G-scope
G-scope a été développé au laboratoire de Biologie et de
Génomique Structurales, pour la visualisation et l’analyse de génomes complets
ou de collections de séquences protéiques.
Pour analyser un génome, G-scope détecte les protéines dans
la séquence d’ADN du génome étudié. Il effectue automatiquement des recherches
dans les banques de données, crée des fiches d’informations sur chaque protéine
étudiée, et calcule plusieurs statistiques avec des sorties graphiques. Chaque
étape peut être validée par l’utilisateur. A l’aide de cet ensemble de données,
le biologiste peut analyser les protéines, leurs relations et leurs
localisations. Il peut valider certaines hypothèses, demander de nouvelles
recherches, affiner les alignements, effectuer une analyse phylogénétique, etc.
G-scope permet ainsi de générer une base de données sous forme de fichiers. A
chaque protéine correspond par exemple un fichier TblastN (contre les séquences
nucléiques des autres génomes complets ), un fichier BlastP (contre les banques
SwissProt, Trembl, SptNew) contenant les protéines homologues, un fichier
d’alignement multiple d’un sous-ensemble de ces protéines homologues, etc.
G-scope est également un programme de visualisation : à
partir d’une vue synthétique du génome ou de la collection étudiée,
l’utilisateur se déplace à l’aide de la souris dans la base de données générée.
Tous les fichiers associés à une protéine donnée sont facilement accessibles
depuis chaque fenêtre, et sont affichés de manière interactive. De nouvelles
recherches ou d’autres programmes peuvent être lancés facilement.
Nous devons ici analyser une collection de séquences de
clones et une collection de séquences de gènes, ceux des puces
Affymetrix : il convient donc de donner à G-scope les fonctionnalités
supplémentaires liées à l’analyse de séquences nucléiques. Si la partie
visualisation du programme peut être réutilisée sans changement majeur, la
partie création de base de données doit s’adapter à un protocole d’analyse de
séquences différent, faisant appel à des programmes tels que BLAST,
RepeatMasker et GenScan.
2.2.2.2
BLAST
Ce programme utilise l'algorithme de Altschul et al. (J.Mol.
Biol. 215; 403-410 (1990)) pour rechercher les segments similaires entre une
séquence donnée (séquence "query") et l'ensemble des séquences
présentes dans une banque nucléique ou protéique. Les séquences de la banque
sont classées en fonction d'un score calculé par BLAST en fonction de
l'homologie avec la séquence query, de la taille de la banque, etc. : l'
"expect". L'homologie entre la séquence query et une séquence de la
banque est d'autant plus significative que l'expect associé est faible.
BLAST regroupe en fait différents sous-programmes selon la
nature de la séquence query et de la banque (cf. [8]) :
|
Sous-programme
|
Type de
séquence en entrée (query)
|
Type de banque
|
Remarque
|
|
BlastP
|
Protéique
|
Protéique
|
|
|
BlastN
|
Nucléique
|
Nucléique
|
|
|
TblastN
|
Protéique
|
Nucléique
|
Les séquences nucléiques de la base sont automatiquement
traduites dans les 6 phases de lecture avant la recherche de segments
homologues. Ceci est donc équivalent à 6 BlastP.
|
|
BlastX
|
Nucléique
|
Protéique
|
La séquence entrée est traduite dans les 6 phases de
lecture avant la recherche de segments homologues. Ceci est donc équivalent à
6 BlastP.
|
|
TblastX
|
Nucléique
|
Nucléique
|
La séquence entrée et les séquences de la banque sont
traduites dans les 6 cadres de lecture avant la recherche de segments
homologues. Ceci est donc équivalent à 36 BlastP.
|
2.2.2.3
RepeatMasker
Des études directes sur
l'ADN ont montré que chez les organismes supérieurs, certaines séquences
nucléotidiques apparaissent de nombreuses fois dans le matériel génétique. Les
génomes eucaryotes, et donc en particulier le génome humain, contiennent en
effet une grande proportion d’ADN répétitif de type « Tandemly repeated
DNA » (séquences répétées souvent associées à des syndromes de maladie),
ou « Interspersed repetitive DNA » (cf. [3]). On estime qu’un tiers
du génome humain est constitué de séquences d’ADN répétitives de type
« Interspersed », qui sont des copies dégénérées d’éléments
transposables : il s’agit d’éléments instables de l’ADN capables de migrer
en différents endroits du génome. Ces séquences répétées ne sont pas
regroupées, mais dispersées en de nombreux endroits du génome. Il existe
différents types de séquences répétées de type Interspersed : LINEs,
SINEs, éléments transposables avec LTR (Long Terminal Repeats), DNA
Transposons.
RepeatMasker est un programme qui détecte les séquences
répétées de type Interspersed, en comparant la séquence soumise à des
librairies de séquences répétées. Les fichiers de sortie du programme sont
d’une part une annotation détaillée des séquences répétées présentes dans la
séquence soumise, et d’autre part une version modifiée de cette séquence, dans
laquelle les séquences répétées détectées sont remplacées par des caractères N.
2.2.2.4
GenScan
GenScan détecte les gènes présents dans la séquence
nucléique qui lui est soumise, en utilisant un modèle probabiliste des
propriétés de structure et de composition des gènes de l’organisme étudié. Le
fichier de sortie donne la liste des gènes prédits, avec leurs exons et
introns, ainsi que les séquences protéiques correspondantes. Contrairement à la
plupart des autres programmes de prédiction de gènes actuellement disponibles,
GenScan traite le cas le plus général : la séquence soumise peut ne pas
contenir de gènes, ou bien en contenir un, ou plusieurs, complets ou partiels,
sur un seul ou sur les deux brins d’ADN. Il reste cependant quelques
restrictions : les unités de transcription sont supposées être non
chevauchantes, et seuls les gènes codant pour des protéines sont considérés.
Les gènes des ARN de transfert et des ARN ribosomaux, par exemple, ne sont donc
pas considérés. Ces gènes n’interviennent pas en principe dans notre étude,
puisque les ARNt et les ARNr n’ont pas de queue polyA, et que seuls les ARN
ayant une queue polyA (ARNm) subissent en principe la transcription inverse au
cours de l’expérience de Differential Display.
2.2.3.1
Collection des clones (Voir
en annexe le déroulement du protocole sur un exemple)
Ces différents programmes sont utilisés dans un protocole
spécifiquement élaboré pour des séquences nucléiques de type cDNA, devant
fournir les deux caractéristiques des gènes dont les clones sont issus, à
savoir leur localisation chromosomique et la fonction de la protéine dont ils
dirigent la synthèse. Tout le déroulement du protocole décrit ci-après a été
automatisé dans le programme G-scope.
La première étape est de localiser les clones sur le génome humain. Or des séquences
répétées peuvent être présentes dans les ARNm et donc dans les clones à
analyser. Il est donc nécessaire de ne pas tenir compte de ces séquences
répétées lorsque l’on compare les clones au génome humain grâce à BlastN, de
sorte à éviter des correspondances multiples non informatives. Pour cela, on
soumet préalablement les séquences des clones à RepeatMasker.
Une fois les séquences répétées maquées, on peut rechercher
la localisation des clones sur le génome humain, en soumettant au programme
BlastN la séquence de sortie de RepeatMasker et en choisissant comme banque de
comparaison celle du génome humain.
Plusieurs cas se présentent alors :
·
aucune correspondance n’est trouvée dans le génome
humain .
·
une seule correspondance est trouvée dans le génome
humain, sans ambiguïté : on connaît alors le chromosome et, au sein de ce
chromosome, le contig d’où provient le clone. Cependant, le génome humain
n’étant pas annoté, on ne connaît pas encore la fonction du gène d’où provient
le clone.
·
plusieurs correspondances sont trouvées, mais l’une est
meilleure que les autres en terme d’expect : on retient comme localisation
le chromosome et le contig de la meilleure correspondance, et on signale le
risque que notre clone provienne d’un gène présent en plusieurs exemplaires
homologues dans le génome.
·
plusieurs correspondances sont trouvées, avec le même
expect : ce cas n’a pas encore été traité.
La seconde étape consiste à déterminer la fonction des gènes dont les clones proviennent. Pour
cela, plusieurs méthodes sont mises en œuvre en parallèle :
·
recherche des homologues de la séquence de sortie de
RepeatMasker dans GenEmbl
grâce au programme BlastN
·
recherche des homologues de la séquence de sortie de
RepeatMasker dans les ESTs, également grâce au programme BlastN
·
soumission de la séquence du contig trouvé (grâce au
BlastN contre le génome humain) au programme GenScan. On détermine parmi les
gènes prédits celui qui comprend notre clone, et on soumet la protéine prédite
correspondante au programme BlastP, de sorte à trouver une protéine homologue
décrite dans les banques protéiques. On détermine alors la protéine décrite
dans les banques qui correspond le mieux à notre protéine prédite. Comme les
banques protéiques contiennent elles-même des protéines prédites, on s’assure
que la protéine trouvée dans les banques a été observée biologiquement,
c’est-à-dire que des cDNA (portions d’ARNm) de cette protéine ont été observés.
Pour cela, on recherche les homologues de cette protéine dans les ESTs grâce au
programme TblastN.
·
Si le BlastP précédent n’a donné aucune protéine
homologue dans les banques protéiques, alors on recherche les homologues de la
protéine prédite directement dans les ESTs grâce à TBlastN : il est
possible en effet que la protéine réelle homologue de notre protéine prédite ne
soit pas encore décrite dans les banques protéiques, mais qu’elle le soit dans
les ESTs via des cDNA correspondant à son ARNm.
Certains critères sont mis en
place afin d’évaluer la fiabilité de la fonction finalement trouvée par
cette méthode :
-
on vérifie que notre clone se trouve bien dans la partie 3’ du
gène prédit par GenScan qui lui correspond.
-
on vérifie que la taille de la protéine prédite et celle de
son homologue dans les banques protéiques correspondent : si la différence
de taille est trop importante, on peut supposer que GenScan a mal prédit la
protéine (fusion de deux gènes, exons prédits surnuméraires ou manquants…).
Les trois méthodes doivent finalement donner la même
fonction.
2.2.3.2
Collection des gènes des puces Affymetrix
Pour les gènes des puces Affymetrix, le protocole se limite
à la recherche de la localisation chromosomique. En effet, il ne s’agit pas
comme précédemment de cDNA inconnus puis séquencés, mais de séquences connues
issues des banques de données. On dispose ainsi de leur « accession
number » dans GenEmbl,
et il s’agit en général de séquences dont la définition contient une fonction.
On recherche la localisation chromosomique de ces gènes selon la même méthode
que pour les clones, c’est-à-dire en soumettant les séquences au programme
RepeatMasker, puis en recherchant les séquences homologues de la sortie de
RepeatMasker dans le génome humain à l’aide du programme BlastN.
Il est aisé de comparer les résultats de Differential
Display avec les profils obtenus en Northern Blot, et en Virtual et Reverse
Northern Blot. En effet, les clones sur lesquels les différents Northerns Blots
ont été réalisés sont issus de bandes du gel de Differential Display. On peut
donc comparer ces profils directement grâce à une requête SQL exécutée avec
Access.
Au contraire, les gènes des puces Affymetrix sont
indépendants des bandes de Differential Display et des clones qui en sont
issus. On ne peut donc pas directement comparer les différents types de profils
d’expression : il faut pouvoir établir une correspondance entre les clones
et les gènes des puces Affymetrix. Cela est possible grâce aux localisations
chromosomiques déterminées lors de l’analyse de séquences. On considère en
effet qu'un clone correspond à un gène des puces Affymetrix s’ils sont
localisés au même endroit sur le génome humain. En pratique, on dispose, grâce
au BlastN contre le génome, de plusieurs localisations possibles (expect
inférieur à 10-3) de part et d'autre. Ce sont les deux ensembles de
localisations possibles qui sont pris en compte pour déterminer un lien
éventuel entre un gène Affymetrix et un clone : il y a lien s'il existe un
couple de localisations chevauchantes parmi tous les couples de localisations
possibles. L'établissement de ces liens est assuré par une procédure spécifique
écrite en TCL dans le code de G-scope, et les liens obtenus sont ensuite
renseignés dans la base de données Access (table "Overlaps").
Remarque : il aurait également été possible de
constituer une banque de données avec l’ensemble des 12 650 séquences des gènes
des puces Affymetrix, et de rechercher les homologues du cDNA parmi ces
séquences grâce au programme BlastN. Mais les résultats en sortie de Blast
n’auraient pas été exploitables, puisque le critère le plus utilisé pour
classer les homologues trouvés, à savoir l’expect, dépend de la taille de la
banque. Le nombre de séquences (12 650) dans la banque des gènes des puces
Affymetrix est trop faible pour que l’on obtienne des expects significatifs.
L’outil informatique développé a permis de réaliser l’analyse
automatique des 2 206 séquences contenues dans la base de données, et de
regrouper toutes les données biologiques pour une exploitation facilitée.
Sur les 2 206 clones séquencés traités avec G-scope, 190
contiennent une séquence répétée détectée puis masquée par RepeatMasker. 31 de
ces clones sont masqués à plus de 90 %, et ne contiennent donc pas assez
d'information pour être soumis à la suite de l'analyse.
Les 2 175 séquences qui n'ont pas été totalement masquées
ont été soumises au programme BlastN de sorte à rechercher leurs homologues
dans le génome humain.
Pour 362 (soit 17 %) de ces clones, aucune correspondance
n'a été trouvée dans le génome humain. Ces clones peuvent :
·
correspondre à des parties du génome humain pas encore
séquencées.
·
provenir de la transcription de l'ADN mitochondrial :
en effet, les mitochondries (organites producteurs d'énergie de la cellule)
contiennent également de l'ADN, qui peut être transcrit et traduit.
·
provenir d’une contamination par la souche bactérienne E. Coli utilisée dans le protocole,
ou encore par des micro-organismes présents dans les tissus prélevés chez les
patients.
1 018 clones sont localisés sans ambiguïté (une seule
localisation possible).
786 clones présentent plusieurs localisations possibles.
Parmi ceux-ci, 222 ont au moins deux localisations indiscernables (même expect)
: ces clones peuvent correspondre à des gènes présents en différents
exemplaires sur le génome. La fonction des gènes correspondant à ces 222
séquences n'a pas encore été recherchée.
La méthode de recherche de fonction la moins fiable semble
logiquement être celle comprenant une étape de prédiction de protéine. En
effet, pour 929 clones, on ne dispose pas de protéine prédite, souvent parce
que la zone du génome où le clone est localisé est située en dehors des gènes
prédits par le programme de prédiction GenScan. De plus, pour 112 clones, on
dispose bien d'une protéine prédite, mais celle-ci n'a pas d'homologue humaine
dans les banques de séquences protéiques. La protéine peut être correctement
prédite, et dans ce cas, la protéine réelle correspondante n'est pas encore
référencée dans les banques. Mais il est également probable que la séquence de
la protéine prédite soit erronée.
Au contraire, les deux autres méthodes (comparaison directe
de la séquence du clone à celles contenues dans les banques GenEmbl et ESTs)
fournissent en général des séquences humaines homologues. Mais d'autres
problèmes se posent : d'une part, la fonction des séquences homologues trouvées
n'est pas toujours référencée, et d'autre part, dans la banque des ESTs
notamment, le nombre de séquences homologues est très élevé, rendant difficile
l'exploitation directe du fichier de sortie généré par le programme BlastN.
Grâce à une procédure écrite en VBA dans le code associé à
la base de données, on peut vérifier la cohérence des fonctions ou définitions
obtenues pour une même séquence par les différentes méthodes. On trouve ainsi
que les définitions trouvées par prédiction de protéine et recherche
d'homologues dans les banques protéiques, par recherche directe dans GenEmbl et
par recherche directe dans la banque d'ESTs, ne sont cohérentes que pour
environ 6 % des séquences analysées.
Cela est dû en partie à la fréquence des échecs lors de la
prédiction de protéine : il arrive que la séquence étudiée ne se trouve pas
dans un gène prédit, auquel cas il est impossible de rechercher les homologues
dans la banque protéique et de disposer d'une définition. En effet, si l'on ne
tient pas compte de la définition (ou l'absence de définition) donnée par cette
méthode, et donc si l'on ne compare que les définitions trouvées dans la banque
des ESTs et dans GenEmbl, alors on obtient une cohérence pour environ 14 % des
séquences.
Un autre facteur responsable de cette faible proportion de 6
% est le fait que l'on ne compare que les définitions stockées dans la base de
données, c'est-à-dire pour chaque méthode celle du meilleur homologue humain.
Il serait possible d'augmenter la proportion de séquences pour lesquelles les
méthodes convergent en prenant en compte pour chaque méthode un ensemble de
bons homologues humains.
Enfin, cette faible proportion est également due au fait que
les définitions des banques protéiques comprennent généralement la fonction du
gène, alors que ce n'est pas toujours le cas dans GenEmbl et dans la banque
d'ESTs.
Le fait que l’ensemble des données soient regroupées dans
une même base et sous le même format permet une analyse globale du projet, ce
qui était beaucoup moins aisé avec différents fichiers issus de différents
programmes. Quelques chiffres concernant la série d'expériences
"Differential Display et Northerns", obtenus par une simple
interrogation de la base par des requêtes SQL, illustrent cette
possibilité d’analyse globale :
·
1 275 bandes ont été sélectionnées (en raison d’un
profil d’expression présentant des différences parmi les types d’échantillons)
parmi l’ensemble des bandes observées sur les gels de Differential Display
comparant l’expression des gènes issus d’échantillons de type E, NE, U, NU, S,
ou NS. Les types de profils d'expression (tumoral, sain ou complexe)
obtenus pour ces bandes se répartissent comme le montre le graphique suivant.
Ce graphique présente également à titre d'exemple la répartition des
spécificités vis-à-vis d'un ou deux types de cancer, pour les profils de type
complexe :
Type
de profil d'expression des bandes sélectionnées
|
|
Spécificité
d'expression des bandes "complexes" vis-à-vis du type de cancer
|
|
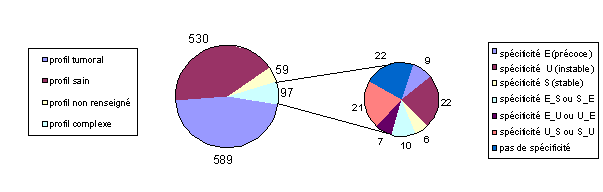
Figure 10
: Profils d'expression des bandes de Differential Display
On voit que les bandes de profil
"complexe" représentent une proportion relativement mineure : les
bandes sélectionnées par les expérimentateurs correspondent donc
majoritairement à des gènes s'exprimant plus dans les cellules tumorales que
dans les cellules saines – ou inversement – pour tous les types de cancers (précoce, stable et instable), et
présentant donc un profil typiquement tumoral – ou sain.
·
3 906 clones, issus de cette sélection de bandes, ont
un profil d'expression en Reverse Northern renseigné ; les profils obtenus se
répartissent comme suit :
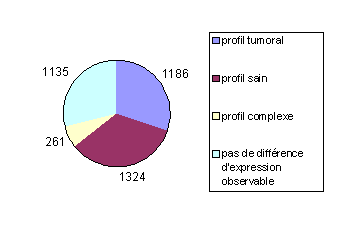
Figure 11
: Profils d'expression des clones en Reverse Northern
On remarque une forte proportion
de clones ne présentant pas de sélectivité vis-à-vis du type de sonde utilisée
(sonde "saine" ou "tumorale"). Or les clones traités en
Reverse Northern proviennent de bandes sélectionnées par les expérimentateurs,
donc présentant pour la majorité une sélectivité d'expression vis-à-vis du
caractère sain ou tumoral des cellules. En mettant en évidence cette
incohérence apparente, la base de données construite devient ici un outil
permettant au biologiste d'évaluer le protocole mis en œuvre et la validité des
profils obtenus.
·
2 206 clones jugés intéressants par leur profil en
Differential Display et en Reverse Northern ont été séquencés, mais il n'existe
parmi ces séquences que 2 002 séquences différentes. En effet, plusieurs clones
peuvent provenir d'une même bande de Differential Display,
alors que ces bandes peuvent ne contenir qu'un seul cDNA : les 4 ou 8 clones issus d'une telle bande
seront donc identiques. On introduit donc souvent une redondance dans les
séquences des clones.
·
226 clones présentant un profil d'expression
intéressant en Differential Display et/ou en Reverse Northern ont été traités
en Virtual Northern, afin de confirmer ce profil. Les profils d'expression
obtenus se répartissent comme suit :
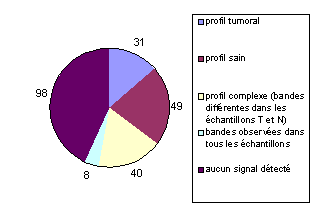
Figure 12
: profils d'expression des clones traités en Virtual Northern
On remarque la forte proportion
de clones ne fournissant pas de signal significatif. Ici encore, le
regroupement des données des différents expérimentateurs, grâce à la base de
données, permet de mettre en évidence les étapes du protocole où des problèmes
auraient pu survenir.
·
142 bandes ont pour l’instant été sélectionnées parmi les
bandes observées sur les seconds gels de Differential Display, c'est-à-dire les
gels permettant de comparer l’expression des gènes dans des cellules de type R
ou NR. La base de données construite ne contient pas encore tous les résultats
de cette seconde expérience de Differential Display, et devra s'adapter à ces
nouvelles données ainsi qu'à celles des expériences qui suivront. Ce point
montre la particularité d'une base vouée à la gestion de données expérimentales
: sa structure doit évoluer en permanence en fonction des protocoles
biologiques.
La base de données construite permet également de comparer
directement les profils obtenus en Differential Display, Reverse Northern et
Virtual Northern. A titre d'exemple, mentionnons que 1 874 clones, soit 48 %
des clones traits en Reverse Northern,
ont des types de profils d'expression (tumoral, sain ou complexe)
cohérents en
Differential Display et en Reverse Northern. Mais l'expérience de Virtual
Northern ne confirme le type de profil que pour 30 clones, soit 13 % des clones
traités en Virtual Northern. Parmi ces 30 clones, 15 ont un profil de type
tumoral, c'est-à-dire que s'expriment sélectivement dans les cellules
tumorales, et 15 ont un profil de type sain.
Il est également possible – et plus intéressant – de
comparer les profils d'expression cités ci-dessus avec ceux obtenus par la
première expérience Affymetrix, mais cette comparaison nécessite d'avoir établi
une correspondance entre les clones et les gènes représentés sur les puces
Affymetrix. Il a été
trouvé au moins une correspondance avec un gène Affymetrix sélectionné par
l'ANOVA pour 753 clones, soit 34 % des clones séquencés (donc dont la
localisation chromosomique a été recherchée par G-scope). Cette proportion
pourrait être augmentée en considérant l'ensemble des gènes présents sur les
puces Affymetrix, mais seuls les gènes sélectionnés par l'ANOVA ont été traités
en "clustering", acquérant ainsi un profil d'expression au même
format que celui obtenu en Differential Display.
3.2.2.1
Comparaison des types généraux de profils
Pour 67 % des clones ayant une correspondance avec un gène
Affymetrix (soit 506 clones), le type de profil obtenu en Differential Display
est corroboré par au moins l'un des gènes Affymetrix correspondants.
Mais ce n'est que pour environ la moitié de ces clones (233,
soit 7.5 % des clones traités en Reverse Northern) que le type de profil obtenu
en Differential Display et en Affymetrix est également confirmé par l'expérience
de Reverse Northern. Ces 233 clones se répartissent en deux groupes :
·
101 clones s'expriment plus fortement dans les cellules
tumorales que dans les cellules saines. Parmi ceux-ci, on trouve par exemple
les clones :
§
A 0564H : Sous-unité b3 de la Na+/K+-
ATPase
§
F D3.3 : Histone
H2
§
I A71h5 : Thymine DNA glycosylase spécifique
des erreurs G/T
§
J 234-G : Inhibiteur de sérine (ou cystéine)
proteinase (nexine)
§
I
A68o6 : Mitosine
§
I
C74a3 : Lysine hydroxylase
§
J 221-A : Récepteur de l'urokinase
§
J 260-D : Transporteur de glucose 3
·
132 clones s'expriment plus fortement dans les cellules
saines que dans les cellules tumorales. Parmi ceux-ci, on trouve par exemple
les clones :
§
A 1252B : Glycoprotéine G1 riche
en proline de la salive
§
F B1.4 :
b-(1,3/1,4)-fucosyltransférase
FT3B
§
CW 44-45 : Hémoglobine
§
I
A69e5 : Glutathione S-transférase A2
§
I G80d6 : Protéine NDP52 du domaine nucléaire
10
§
J 140-A : Kératine 13
§
J 237-F : Calgizzarine (protéine S100C, MLN
70)
§
PM 98g : Leucotriène A-4 hydrolase
On remarque qu'aucun clone ne présente un profil complexe à
la fois en Differential Display, en Reverse Northern et en Affymetrix.
L'expérience de Virtual Northern ne confirme quant à elle le
type de profil obtenu par les trois autres méthodes que pour 9 de ces 233
clones, 3 ayant un profil tumoral et 6 ayant un profil sain. Ainsi, la base de
données met ici en évidence la difficulté à établir avec certitude des profils
d'expression génique : les quatre méthodes mises en œuvre ne concordent au niveau
du type de profil que pour une infime proportion du nombre total de clones.
3.2.2.2
Comparaison des spécificités vis-à-vis du type de cancer
Pour les clones présentant un profil d'expression de type
tumoral, il est possible de comparer la spécificité des profils obtenus
vis-à-vis d'un ou deux types de cancer. Cette spécificité n'est généralement
pas exactement la même en Differential Display et en Affymetrix. Cependant,
pour certains clones, on obtient des spécificités proches.
Par exemple, le clone A 0564H, issu du gène de
l'interleukine 8 (selon l'analyse de séquence), semble s'exprimer plus
fortement dans les cellules tumorales précoces et instables en Differential
Display. Le gène Affymetrix qui lui correspond du point de vue de la
localisation chromosomique et de la fonction,
paraît présenter une plus forte expression dans les cellules tumorales
précoces et stables. La différence apparente de spécificité provient de la
façon dont elle est déterminée : ce sont les deux plus fortes intensités
d'expression qui sont prises en compte, et non la plus forte seulement. Si cela
permet une caractérisation plus fine des profils, leur comparaison n'en est
plus triviale.
On a vu que la recherche de la localisation chromosomique a
échoué pour une proportion assez forte (17 %) des séquences soumises, et que
cela peut être dû au fait que les séquences analysées peuvent provenir de
contaminations bactériennes (E. coli
et autres micro-organismes éventuellement présents dans les tissus des
patients), ou bien du génome mitochondrial.
Pour détecter une contamination éventuelle, une solution
consisterait à comparer les séquences des clones à l'ensemble des génomes bactériens,
et à masquer les parties de séquences identiques à 100 % à une séquence
bactérienne, avant de comparer le clone au génome humain. Cette solution
permettrait de ne considérer que les fragments de séquence humains pour la
suite de l'analyse.
Cependant, lorsque l'on compare la séquence à celles de la
banque GenEmbl afin d'obtenir la fonction associée à la séquence, on peut
également vérifier une provenance étrangère au génome humain : en effet,
GenEmbl ne contient pas seulement des séquences humaines, mais aussi des
séquences issues d'autres organismes, dont certaines bactéries.
On a vu que la recherche de la fonction des clones passe par
trois méthodes, dont deux consistent à rechercher directement les homologues du
clone dans les banques nucléiques GenEmbl et ESTs. Or un problème précédemment
souligné est le nombre trop important de séquences homologues trouvées dans
certains cas.
Le problème vient parfois du fait que la séquence du clone
comparée aux banques contient encore des séquences répétées, malgré le passage
par le programme RepeatMasker en début de protocole. En effet, RepeatMasker
compare la séquence donnée à une librairie de séquences répétitives, et masque
la partie de la séquence soumise similaire à une séquence répétée si cette similarité dépasse un certain seuil, qui
dépend de la longueur de la zone similaire. Or lorsqu'une séquence contient
un fragment de séquence répétée trop
court (la fin d'une séquence "Alu repeat" par exemple), ce seuil
n'est pas atteint, et la séquence n'est pas masquée. Lorsqu'on compare cette
séquence à une banque nucléique, toutes les séquences de cette banque contenant
la séquence répétée seront données comme homologues.
Une solution consisterait à soumettre à RepeatMasker le
contig du génome humain sur lequel est localisé le clone. La séquence du contig
est suffisamment longue pour contenir les séquences répétées entières : toutes
ces séquences répétées sont masquées puisque le seuil pourra être atteint. Il
faudrait ensuite déterminer si la zone où est localisé le clone est masquée, et
si oui, re-masquer le clone en conséquence. On pourrait ensuite effectuer la
comparaison de cette séquence re-masquée avec les banques nucléiques.
La troisième méthode pour connaître la fonction des clones
consiste à prédire la protéine correspondant au clone (en soumettant le contig
adéquat du génome humain au programme GenScan) et à rechercher ses homologues
dans la banque de séquences protéiques. Du fait de cette étape prédictive,
cette méthode est la moins fiable et n'aboutit pas toujours.
Cependant, les deux autres méthodes fournissent dans de
nombreux cas des résultats peu exploitables, dans la mesure où la fonction des
séquences stockées dans GenEmbl et dans la banque d'ESTs n'est pas toujours
renseignée. Il est donc nécessaire d'intégrer une confrontation à la banque
protéique dans le protocole d'analyse. Pour cela, on aurait pu rechercher
directement les homologues des clones dans les banques protéiques grâce à
BlastX, mais cela ne donne en général aucun résultat. En effet, les séquences
soumises correspondent à la partie 3’UTR des ARNm, c'est-à-dire à une partie
non traduite in vivo : la faire traduire automatiquement par le programme
BlastX n'a pas de sens biologique. La méthode prédictive reste donc nécessaire.
Enfin, une partie des séquences n'a pas été traitée en ce
qui concerne la recherche de fonction : il s'agit des clones présentant deux
(ou plus) localisations chromosomiques aussi probables l'une que l'autre. Le
problème se pose pour la méthode nécessitant une prédiction de protéines :
lequel des contigs doit-on soumettre au programme de prédiction GenScan ? Dans
la mesure où l'on ne dispose pas de critère pour choisir l'un des contigs que
plutôt que l'autre, la solution consisterait à les traiter tous. On obtiendrait
alors plusieurs protéines prédites pur le même clone, qu'il faudrait comparer
entre elles (grâce au programme FastA par exemple). Si elles sont semblables,
alors notre clone correspond à un gène présent en plusieurs homologues (dits
paralogues) dans le génome, et on peut continuer le protocole (BlastP contre la
banque protéique, puis TblastN contre la banque d'ESTs). Si au contraire, elles
sont très différentes, alors poursuivre cette méthode n'a pas de sens :
GenScan a vraisemblablement mal prédit au moins une des protéines, mais on ne
sait pas laquelle.
L’analyse globale des profils d’expression génique rendue
possible par le regroupement des données nécessite cependant un certain nombre
d’impératifs pour être rigoureuse. Tout d’abord, la saisie des profils
d’expression par les différents expérimentateurs doit être la plus homogène
possible du point de vue des notations. Une limitation des types de profils
valides doit pour cela être mise en place. De plus, le calcul de proportions
relatives aux clones peut être faussé par le fait que certains expérimentateurs
ont entré tous les clones dans la base, même si certains avaient la même
séquence, alors que d’autres n’y ont entré que les séquences uniques.
Par ailleurs, s’il est possible d’obtenir et de comparer
simplement et automatiquement (par des requêtes SQL) les profils d’expression,
il faut tenir compte du fait que ces profils doivent dans certains cas être
établis qualitativement : en Differential Display par exemple,
l’expérimentateur doit évaluer l’intensité des bandes. Il y a plusieurs
expérimentateurs, donc il y a potentiellement un risque de comparaisons
erronées.
Une fois ces précautions prises, l’outil développé peut être
utilisé pour déterminer quelles protéines sont sur- ou sous-exprimées dans les
différents types de cancers des VADS. Un tri manuel de ces protéines s’avère
ensuite nécessaire. En effet, il faudra par exemple distinguer parmi les
protéines surexprimées dans les cellules tumorales :
·
celles dont la surexpression est une conséquence de la
division accélérée des cellules (protéines ribosomales, mitosine, …)
·
celles dont la surexpression peut être la cause de
cette division accélérée et de l'état tumoral (protéines impliquées dans la
régulation du cycle cellulaire par exemple). Ces protéines sont bien sûr les
plus intéressantes dans le cadre de la recherche de cibles thérapeutiques.
La base construite est vouée à la gestion de données
expérimentales, et doit donc s'adapter à la progression du protocole
biologique. Deux nouvelles expériences sont en cours et leurs résultats devront
être intégrés à la structure existante :
·
une nouvelle expérience de Differential Display est
menée pour caractériser l'expression des gènes dans les tissus de patients
répondant à la chimiothérapie (notés R) et dans ceux de patients pour lesquels
cette thérapie est inefficace (notés NR).
·
une nouvelle série d'expériences de puces à ADN
Affymetrix est réalisée, avec des échantillons qui correspondent à de nouveaux
patients. Les résultats obtenus devront être intégrés à ceux de la première
série.
Le code VBA intégré doit également évoluer. En effet, la
base de données doit à terme être utilisée par tous les partenaires du projet,
et non par l'IGBMC seulement. Une
interface Internet doit donc être développée dans ce but.
Par ailleurs, le mode de dialogue de l'application Access
avec G-scope peut être amélioré. Ce dialogue a en effet été réalisé par
l'intermédiaire de fichiers ; une possibilité d'amélioration serait un dialogue
direct, où G-scope (ou bien sa version Internet, W-scope) serait lancé et
interrogé depuis la base de données.
La solution informatique retenue pour répondre aux besoins
de l’équipe du Dr. Wasylyk, alliant une base de données relationnelle à un
programme dédié à l’analyse de séquences, a permis de regrouper l’ensemble des
données biologiques acquises jusqu’à présent, ainsi que d’analyser
automatiquement la majeure partie des séquences disponibles.
Cette solution permet une exploitation plus globale des
résultats biologiques obtenus, et ce à deux niveaux : l’exploitation peut
se faire, au niveau d’une expérience, pour l’ensemble des gènes traités par les
différents expérimentateurs, mais aussi au niveau d’un gène, pour l’ensemble
des expériences.
Les deux modules de cette solution sont amenés à
évoluer : l’analyse de séquences peut être affinée, et la base de données
doit notamment s’orienter vers l’Internet.
Ce stage a été pour moi l’occasion de mettre en pratique la
création d’une base de données, et de me familiariser avec les techniques
d’étude de l’expression génique, avec l’analyse de séquences, et avec un
nouveau langage de programmation (TCL).
J’ai également découvert le travail des bioinformaticiens en
partenariat avec les biologistes, et par là même les enjeux d’un tel
partenariat. En effet, la bioinformatique permet ici aux biologistes une
gestion facilitée de leurs nombreuses données. Mais rappelons également que
l’analyse de séquences, menée en partenariat avec l’équipe de bioinformatique
du Laboratoire de Biologie et de Génomique Structurales, a donné lieu à
l’élaboration d’un protocole d’analyse de séquences nucléiques humaines, ce qui
représente une étape importante pour l’un des axes de recherche de cette
équipe, à savoir l’étude des génomes eucaryotes.
J’ai enfin été confrontée à certains défis, comme la
nécessité de maîtriser deux disciplines donc deux langages, ou encore le
travail avec une définition floue et évolutive des besoins.
Glossaire
(cf. [4],
[5], [6])
ADN
Abréviation d'Acide DésoxyriboNucléique. Macromolécule
servant de support de l'information génétique chez la plupart des êtres vivants
(la seule exception connue étant les virus à ARN). L'ADN se trouve dans presque
toutes les cellules. Il est aussi présent dans certains organites cellulaires
(chloroplastes et mitochondries). Chez les procaryotes, il est dans le cytoplasme ;
chez les eucaryotes, il est dans un organite spécialisé, le noyau cellulaire.
Sa structure est universelle, seule la longueur de la molécule variant selon
les espèces. Constituant des chromosomes et de la chromatine, l'ADN est le
support matériel de l'information et du patrimoine génétiques.
Un brin d'ADN est constitué d'une chaîne plus ou moins
longue de désoxyribonucléotides (nucléotides comportant un sucre, le
désoxyribose, et une base azotée choisie parmi la thymine T, la cytosine C,
l'adénine A et la guanine G). Chaque base est appariée à une autre, dite
complémentaire, par des liaisons hydrogène, suivant deux couples possibles :
A--T et G--C.
L'ADN est constitué de deux brins. Chaque base d’un brin est
appariée à une base de l’autre brin qui lui est complémentaire. Les deux brins
reliés complémentaires sont arrangés en double hélice.
ADN complémentaire (ADNc ou cDNA)
ADN simple brin synthétisé à partir d’un brin d’ARN :
il est obtenu après une réaction de transcription inverse d'un ARN matur et
représente ainsi la copie de l'ARN. En biologie moléculaire, cette synthèse
permet d’obtenir des copies d’ARN messager sous forme d’ADN : l'ADNc offre
l’avantage d'être plus stable que la molécule d'ARNm et de pouvoir être stocké,
copié et séquencé.
ARN
Abréviation de Acide RiboNucléique. Acide nucléique à
structure proche de celle de l'ADN, qui joue un rôle capital dans la synthèse
des protéines et constitue le support de l'information génétique chez certains
virus. Le sucre présent dans les nucléotides de l'ARN est le ribose ; les
quatre bases azotées sont l'adénine A, la cytosine C, la guanine G, l'uracile U
remplaçant la thymine T présente dans l'ADN, dans les appariements avec A.
L'ARN est moins stable que l'ADN, et son rôle est généralement celui d'une
molécule transitoire, à durée de vie relativement courte. L'ARN est constitué
d'un seul brin, ce qui lui permet de présenter de nombreuses structures
(boucles, doubles chaînes), qui jouent un rôle important dans la régulation de
la traduction. En biologie moléculaire, on utilise l'enzyme appelée
transcriptase inverse pour copier l'ARN en ADN (dit ADNc ou cDNA) afin de
pallier le peu de maniabilité de l'ARN.
On rencontre différents types d'ARN dans les cellules (ARN
messager ou mRNA, ARN de transfert ou tRNA, ARN ribosomal ou rRNA).
Voir Transcription, Traduction.
ARN messager (ARNm ou mRNA)
Molécule d'ARN (comprenant entre 1000 et 50000 bases),
produite par la cellule à partir de l'A.D.N. lors de la transcription, à
l'origine de la matrice sur laquelle viennent se synthétiser les protéines. La
maturation éventuelle ou épissage des pré-ARNm issus de la transcription leur
permet d'acquérir une structure fonctionnelle. La séquence des bases azotées
portées par les nucléotides d'un ARNm porte le code nécessaire à la synthèse
d'une protéine. La lecture de ce code est effectuée par le ribosome au cours de
la traduction. Les ARNm sont éliminés
très rapidement et ne représentent en moyenne qu'un pour cent de la masse des
ARN d'une cellule.
Biopsie
Prélèvement
sur le corps vivant d'un fragment de tissu ou d'organe en vue d'un examen
microscopique ou d'une analyse biochimique.
BLAST
Basic Local
Alignment Search Tool
bp
Unité
de mesure de la taille d’une séquence d'ADN en paires de bases. kb : kilo paires
de bases = 1000 bp.
Cancer
Tumeur maligne formée par la multiplication désordonnée de
cellules.
cDNA
Voir ADN complémentaire.
Champ
Colonne d'une table. Ensemble des valeurs qui correspondent
à une même propriété (exemple : champ Séquence de la table Clones). Toutes les
données d'une colonne sont de même type.
Clé primaire
Propriété ou ensemble de propriétés (colonne ou ensemble de
colonnes) identifiant une ligne de façon unique. Par définition, la clé
primaire doit donc avoir une valeur différente pour chaque ligne.
Contig
Séquence obtenue en joignant une collection de séquences
chevauchantes. Dans le cas du génome humain, les contigs sont des fragments de
chromosome contenant des séquences (gènes ou non) dans un ordre connu. Mais la
localisation précise de ces contigs sur le chromosome et leur position relative
n'est pas forcément connue.
DATAS
Differential Analysis of Transcripts with Alternative
Splicing : analyse des exons et introns, ou des insertions et délétions dans
une population de mRNA.
DNA
cf. ADN
DNA transposons
Ces séquences répétées contiennent le gène de la
transposase, sans intron, entouré de deux courtes séquences répétées inversées.
Eléments transposables avec LTR (Long Terminal Repeats)
Ces séquences répétées ont une longueur allant de 1.5 à 10
kb. Contrairement aux LINEs et aux SINEs,
elles contiennent le gène de la transcriptase inverse (cf. Transcription
inverse), entouré de séquences repeats dites terminales de 300 à 1000 bp. Cette
catégorie de séquences répétées comprend les rétrovirus endogènes humains non
fonctionnels (hERV).
Enzyme
Protéine produite par un être vivant pour catalyser des
réactions biochimiques spécifiques, dans des conditions compatibles avec la
vie.
Epissage – Epissage alternatif
Phénomène de maturation des ARN messagers chez les
eucaryotes : certaines parties sont "découpées" (excisées) après la
transcription et non exprimées lors de la traduction (ce sont les introns). Les
autres parties (exons) sont recollées (épissées) et les bases azotées de leurs
nucléotides codent effectivement pour la protéine exprimée. Lors de l'épissage,
certains exons peuvent être enlevés avec les introns, on parle alors d'épissage
alternatif puisque un même gène peut donner naissance à plusieurs ARN messagers
avec des combinaisons d'exons différentes.
EST
Expressed Sequence Tag. Une séquence EST est une étiquette
(fragment d'une extrémité) d'un ADNc. Une séquence EST est donc une séquence de
100 à 150 nucléotides d'ADNc correspondant à une des extrémités d'un ARNm.
Eucaryote
Etre vivant dont le matériel génétique de chaque cellule est
enfermé dans un noyau limité par une double membrane (champignons, levures,
animaux, végétaux). L'existence de ce noyau cellulaire donne son nom aux acides
nucléiques comme l'ADN et l'ARN.
Voir Procaryote.
Exérèse
Ablation chirurgicale d'un tissu ou organe inutile ou
nuisible à l'organisme, ou d'un corps étranger.
Exon
Partie d'un gène eucaryote qui contient une séquence codante
et qui est susceptible d'être conservée dans l'ARN lors de l'épissage. Chez les
organismes eucaryotes, tout l'ARN. issu de la transcription de la molécule
d'ADN ne se retrouve pas dans les ARN messagers cytoplasmiques (V. cytoplasme).
L'épissage élimine des séquences non codantes (introns) et joint les séquences
codantes (exons) bout à bout.
Voir Intron.
Gène
Le gène correspond à un fragment de la molécule d'ADN, une
séquence de nucléotides, qui comprend un promoteur de transcription suivi d'une
séquence codant pour un ARN. Cet ARN peut avoir une fonction biologique ou
coder pour une protéine. Par l'intermédiaire de ces protéines (enzymes,
récepteurs, canaux, anticorps…), les gènes déterminent les caractéristiques et
propriétés de l'organisme (son immunité, sa croissance, etc...). Certaines
parties des gènes (les introns) ne sont pas exprimées en protéines lors de la
traduction (car ils sont éliminés au cours de l'épissage chez les eucaryotes),
les séquences codant la synthèse d'une protéine s'appellent exons. L'expression
des gènes est régulée à différents stades de leur action.
Génome
Totalité du matériel génétique chromosomique d’un organisme.
Hybridation
Appariement entre séquences nucléiques complémentaires par
des liaisons hydrogènes spécifiques, A-T et G-C pour l'ADN ; A-U et G-C pour
l'ARN.
Intron
Partie d'un gène non traduite en protéine parce que la
séquence correspondante est excisée de l'ARN. lors de la maturation en A.R.N.
messager (épissage). Les introns n'existent que chez les eucaryotes. Ils
peuvent constituer la majeure partie d'un gène. Ils contiennent parfois des
séquences régulatrices, mais il est généralement impossible de leur attribuer
un rôle quelconque.
Voir Exon.
LINEs
Long
Interspersed Nuclear Elements. Ils contiennent l’élément L1 (Kpn repeat)
associé à des régions riches en A-T. Présents chez les mammifères, leur
longueur est généralement de 6 à 8 kb. Comme les SINEs, les LINEs peuvent être
transcrits (ils contiennent des promoteurs pour la RNA polymérase III), mais ne
codent pour aucune protéine capable de catalyser une rétrotransposition.
Métastase
Foyer de dissémination secondaire d'une tumeur maligne
primitive se développant généralement à distance de celle-ci.
mRNA
Voir ARN messager.
Nucléotide
Constituant élémentaire des acides nucléiques (ADN ou ARN),
composé d'une base azotée (adénine A, guanine G, cytosine C, ou thymine T dans
l'ADN ou uracile U dans l'ARN), associée à un ou plusieurs phosphates, et à un
sucre (ribose dans l'ARN ou à un désoxyribose dans l'ADN).
PCR
Polymerase Chain Reaction. Le but de cette méthode est de
multiplier en chaîne une petite quantité d'ADN disponible. Elle consiste à
répéter n fois le cycle suivant :
(i)
la séparation des deux brins d'ADN grâce à une température
élevée (environ 95°C) afin d'obtenir des molécules d'ADN monobrin
(ii)
l'hybridation d'oligonucléotides (amorces) complémentaires
d'une séquence de l'ADN monobrin à amplifier (la température est alors ramenée
à une valeur comprise entre 40°C et 65°C afin de permettre une bonne
hybridation des amorces)
(iii)
la réaction de synthèse du brin complémentaire par une ADN
polymérase thermostable (la Taq Polymerase) à partir des oligonucléotides,
réalisée à la température optimale de 72°C.
(i)
les deux brins d'ADN sont séparés grâce à une température
élevée, etc.
Procaryote
Se dit des cellules dépourvues de noyau cellulaire. Les
êtres vivants procaryotes sont généralement unicellulaires comme les bactéries,
les cyanobactéries, les archéobactéries. Leur matériel génétique est de l'ADN
circulaire diffus dans le cytoplasme de la cellule.
Voir Eucaryote.
Protéine
Macromolécule organique composée essentiellement d'acides
aminés reliés par la liaison peptidique. Seuls vingt acides aminés entrent dans
la composition des protéines naturelles. Les protéines interviennent dans
toutes les réactions biochimiques des organismes, notamment grâce à leur
structure spatiale. Une protéine est l'expression d'un gène qui permet sa
synthèse au sein des cellules, au cours du processus de traduction des ARN
messagers.
Ribosome
Complexe présent dans le cytoplasme de la cellule, constitué
de plusieurs parties protéiques et d'ARN ribosomal. Les ribosomes interviennent
dans la synthèse des protéines. Ils effectuent, avec l'aide des ARN de
transfert, la traduction en protéines des ARNm, sur lesquels ils s'accrochent.
RNA
Voir ARN.
RT-PCR
Reverse Transcription Polymerase Chain Reaction.
Amplification par PCR de cDNA issus de transcrption inverse d’ARN messagers.
Séquence peptidique
Ordre des acides aminés sur la chaîne d’acides aminés
formant une protéine. Chaque acide aminé est représenté par une lettre (V pour
valine, L pour leucine…)
Séquence nucléique
Ordre des bases sur la chaîne linéaire de nucléotides
formant un acide nucléique (ADN ou ARN) : chaque nucléotide étant
représenté par l’initiale de la base qui le constitue (T pour thymine, C
pour cytosine, A pour adénine et G pour guanine).
SGBD
Système de gestion de bases de données. Ensemble coordonné
de logiciels capable de décrire, mémoriser, manipuler, interroger les ensembles
de données constituant les bases, et capable de gérer la sécurité et la
confidentialité dans un environnement multi-utilisateurs avec des besoins
variés et pouvant interagir simultanément sur ces données.
SINEs
Short
Interspersed Nuclear Elements. Cette famille comprend notamment les
« Alu repeat », nommées ainsi car elles ont été caractérisées grâce à
l’enzyme de restriction Alu. Ces séquences, spécifiques des primates et riches
en G-C, sont les plus abondantes dans le génome humain avec un nombre de copies
supérieur à 106. Elles se trouvent le plus souvent dans les régions
non traduites (introns) et leur fonction est inconnue. Leur longueur est de 280
bp environ. Les SINEs peuvent être transcrits (ils contiennent des promoteurs
pour la RNA polymérase III), mais ne codent pour aucune protéine capable de
catalyser une rétrotransposition.
SQL
Structured
Query Language. Langage de manipulation de bases de données développé
par IBM. C'est un standard industriel.
Table
Suite de lignes, ou enregistrements. Toutes les lignes ont
même structure, ou format.
Traduction
Processus au cours duquel le message génétique des ARN
messagers est traduit en la séquence d'acides aminés codée par cet ARN. Le code
pour chaque acide aminé successif est une combinaison de trois nucléotides
(triplet ou codon) qui sont déchiffrés les uns après les autres au niveau du
ribosome : lors de la traduction, les deux sous-unités du ribosome
s'assemblent à une extrémité d'un ARNm correspondant au début de la séquence
codante. Le ribosome va ensuite progresser de codon en codon (trois nucléotides)
jusqu'à la fin de la molécule d'ARN. A chaque codon, le ribosome s'arrête, et
un acide aminé est ajouté à la chaîne protéique naissante, en fonction du
codon, selon la règle du code génétique.
Transcription
Transfert de l'information génétique d'un gène, depuis une
molécule d'ADN vers une molécule d'ARN.
Ce processus est effectué par l'ARN polymérase, un ensemble
d'enzymes (appelé aussi transcriptase) qui recopie un brin d'ADN en ARN. Les
nucléotides de l'A.R.N. sont assemblés en une chaîne complémentaire de la
séquence de nucléotides de l'A.D.N. (aux bases azotées C, A, G et T de l'ADN
correspondent respectivement les bases A,C,U et G de l'ARN). L'ARN se détache
lorsque le signal de fin de transcription est décodé. Il subit alors plusieurs
maturations (dont la polyadénylation et l’épissage) pour devenir un ARN
messager (ARNm).
Transcription inverse
Action de la transcriptase inverse, enzyme qui fabrique un
brin d'ADN dit ADN complémentaire à partir d’un brin d’ARN. Cette enzyme n'est
trouvée qu'en accompagnement de certains virus dits rétrovirus. Ce
processus, inverse de la transcription normale, est utilisé en biologie
moléculaire pour synthétiser de l’ADN complémentaire des brins d’ARNm.
Tumeur
Augmentation de volume d'un tissu ou d'une partie d'un organe,
due à une multiplication des cellules. Ces tumeurs peuvent être bénigne (sans
gravité) ou malignes (cancers).
UTR
Untranslated
Region. Partie non traduite d'un ARNm située à l'une de ses extrémités
(3' ou 5').
VADS
Voies Aéro-Digestives Supérieures : bouche, pharynx,
fosses nasales, sinus et larynx.
VBA
Visual
Basic for Applications
Références
Encyclopédie
[1]
Encyclopédie® Microsoft® Encarta 97. © 1993-1996 Microsoft Corporation
Sites Internet
Cours de biologie :
[2] http://www.biology.com/learning/transcription/images/euovrvw.gif
[3] http://www.neuro.wustl.edu/neuromuscular/mother/dnarep.htm
Dictionnaires de biologie :
[4] http://www.sciences-en-ligne.com/Dictionnaire/List_themes.asp
[5] http://www.atoute.org/dictionnaire_medical.htm
[6] http://perl.club-internet.fr/cgi-bin/ehmel/ehmel_search.pl?query=biopsie
Techniques de génétique :
[7] http://www.univ-montp1.fr/biotech/Genomique/Genomique_contenu.htm
Banques et analyse de séquences :
[8]
http://www-igbmc.u-strasbg.fr/TUTORIAL/ATRIUM/atrium.html
Autres
Rapport de DEA :
[9] Anne
Cromer, Identification de gènes
différentiellement exprimés dans les cancers des VADS par la technique de
Differential Display, 2000
Annexe
Illustration du protocole
d’analyse de séquences des clones
On étudie par exemple le cDNA humain
dont la séquence est la suivante (format FastA) :
>Homo sapiens cDNA
GTAAGGGTGTACTAGGGGATAGGATGATGTAAGAGAATGAGAAAGATGAC
CAAAAGGTTGGTGGTAGGGAGGCTTTTTCGTTATTTCCAAATACTTGAGA
AATTACCTTTTGGTTTACAAATCTATGATCAACTTATTCCATTAAATAGA
TACATTAAAAAAATTAAAAACTGATTCTTCTGCAGAGCACTGGTGTTTCT
TTTTATAACCCCTTGAAACAAGTCTCTCACCTGAGCCTGTCTAAACTTTC
GGAGGGAGTTTATTATTGAGTCTTTATCTGTGACAGTATTTGGAGATTTA
GGGATTTGATACTTAGGCCTTTGAATTTTAGAATACAAAAAGAGAAGCAA
GCCAGACATGGTGGCTCACACCTGTAATCCCAATACTGGGAAGCCAAGGT
GGGAGTATCGCTTGAGCCCAGGAGTTTGAGACCGACATGGGCAACATGAC
AAGACCCCATCTCTACAAAAAAATTAAAAAATTAGCCAGGCATGGTGGCA
CATGCCTACTCCCAGCTCCCAAGGAGACTGAGATGGGAGGATCCCTGGAG
CCCTGAAGCTTGAGGCTACAGTGAGCCTTGATTGTGTCACTGCACTCCAG
CTTGGGATGAACAGAGACCCCTGTCTCGACGAAATTAAACCCAAAAAAAC
AGAAACAAAAAAAAGAGACGCCGGGGGGACCATAAAAGAAAAAAAACTAA
GAAAAAACTGGGAGCACACACGACCACGGGGGCCAAAGAACGGTAAAACA
TAGACGAACACAAG
RepeatMasker
La
séquence étudiée est soumise à RepeatMasker. Il retourne la séquence soumise où
les séquences répétées sont masquées …
>Homo
sapiens cDNA (masked)
GTAAGGGTGTACTAGGGGATAGGATGATGTAAGAGAATGAGAAAGATGAC
CAAAAGGTTGGTGGTAGGGAGGCTTTTTCGTTATTTCCAAATACTTGAGA
AATTACCTTTTGGTTTACAAATCTATGATCAACTTATTCCATTAAATAGA
TACATTAAAAAAATTAAAAACTGATTCTTCTGCAGAGCACTGGTGTTTCT
TTTTATAACCCCTTGAAACAAGTCTCTCACCTGAGCCTGTCTAAACTTTC
GGAGGGAGTTTATTATTGAGTCTTTATCTGTGACAGTATTTGGAGATTTA
GGGATTTGATACTTAGGCCTTTGAATTTTAGAATACAAAAAGAGAAGCAN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNGAGACGCCGGGGGGACCATAAAAGAAAAAAAACTAA
GAAAAAACTGGGAGCACACACGACCACGGGGGCCAAAGAACGGTAAAACA
TAGACGAACACAAG
…
ainsi qu’un fichier décrivant les séquences répétées détectées :
==================================================
file
name: exseq.txt
sequences: 1
total
length: 764 bp
GC
level: 42.41 %
bases
masked: 315 bp ( 41.23 %)
==================================================
number of length
percentage
elements* occupied
of sequence
--------------------------------------------------
SINEs: 1 315 bp 41.23 %
ALUs 1 315
bp 41.23 %
MIRs 0 0 bp 0.00 %
LINEs: 0 0 bp 0.00 %
LINE1 0 0
bp 0.00 %
LINE2 0 0
bp 0.00 %
L3/CR1 0 0
bp 0.00 %
LTR
elements: 0 0 bp 0.00 %
MaLRs 0 0
bp 0.00 %
ERVL 0 0
bp 0.00 %
ERV_classI 0 0 bp 0.00 %
ERV_classII 0 0 bp 0.00 %
DNA elements: 0 0 bp 0.00 %
MER1_type 0 0 bp 0.00 %
MER2_type 0 0 bp 0.00 %
Unclassified: 0 0 bp 0.00 %
Total
interspersed repeats: 315 bp 41.23 %
Small
RNA: 0 0 bp 0.00 %
Satellites: 0 0 bp 0.00 %
Simple repeats: 0 0 bp 0.00 %
Low
complexity: 0 0 bp 0.00 %
==================================================
*
most repeats fragmented by insertions or deletions
have been counted as one element
The
sequence(s) were assumed to be of primate origin.
RepeatMasker
version 04/04/2000 default
ProcessRepeats
version 04/04/2000
Repbase version 01/04/2001
On voit
que RepeatMasker a masqué 41 % de la séquence du cDNA qui lui a été soumis. La
séquence répétée masquée est du type « Alu repeat ».
BlastN
contre le génome humain
On soumet
la séquence donnée par RepeatMasker au programme BlastN pour rechercher les
séquences homologues dans le génome humain. On a le fichier de sortie suivant
(extrait) :
BLASTN 2.2.1 [Aug-1-2001]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped
BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query=
Homo sapiens cDNA (masked)
(764 letters)
Database:
Human genome
26,786 sequences; 3,085,169,031
total letters
Searching...................................................done
Score E
Sequences
producing significant alignments: (bits)
Value
HS12:NT009471_17
Continuation (18 of 20) of NT009471 from base 1... 607 e-171
HS12:NT009471_16
Continuation (17 of 20) of NT009471 from base 1... 607 e-171
HS02:NT022380
NT_022380 Homo sapiens chromosome 2 working draft ... 46 0.031
HS18:NT024993
NT_024993 Homo sapiens chromosome 18 working draft... 44 0.12
HS18:NT024983
NT_024983 Homo sapiens chromosome 18 working draft... 44 0.12
HS15:NT024731
NT_024731 Homo sapiens chromosome 15 working draft... 44 0.12
HS15:NT010204_12
Continuation (13 of 36) of NT010204 from base 1... 44 0.12
HS09:NT024025
NT_024025 Homo sapiens chromosome 9 working draft ... 44 0.12
HS09:NT023947_2
Continuation (3 of 4) of NT023947 from base 2000... 44 0.12
HS09:NT023947_1
Continuation (2 of 4) of NT023947 from base 1000... 44 0.12
HS09:NT023947_0
NT_023947 Homo sapiens chromosome 9 working draf... 44 0.12
HS05:NT006931
NT_006931 Homo sapiens chromosome 5 working draft ... 44 0.12
HS01:NT022035
NT_022035 Homo sapiens chromosome 1 working draft ... 44 0.12
HS22:NT011520_097
Continuation (98 of 230) of NT011520 from base... 42 0.48
HS19:NT011145_01
Continuation (2 of 12) of NT011145 from base 10... 40 1.9
HS09:NT023967_3
Continuation (4 of 7) of NT023967 from base 3000... 40 1.9
HS05:NT006654_02
Continuation (3 of 16) of NT006654 from base 20... 40 1.9
HS05:NT006617_13
Continuation (14 of 16) of NT006617 from base 1... 40 1.9
HS02:NT005465
NT_005465 Homo sapiens chromosome 2 working draft ... 40 1.9
HS02:NT022114_0
NT_022114 Homo sapiens chromosome 2 working draf... 40 1.9
>HS12:NT009471_17
Continuation (18 of 20) of NT009471 from base
1700001 (NT_009471 Homo sapiens
chromosome 12 working
draft sequence segment. 2/2001)
Length = 110000
Score =
607 bits (306), Expect = e-171
Identities = 341/351 (97%), Gaps = 3/351 (0%)
Strand = Plus / Minus
Query: 1 gtaagggtgtactaggggat-aggatgat-gtaagagaatgagaaagatgaccaaaaggt 58
||||||||||||||||||||
|||||||| ||||||||||||||||||||||||||||||
Sbjct: 3584 gtaagggtgtactaggggattaggatgattgtaagagaatgagaaagatgaccaaaaggt
3525
Query: 59
tggtggtagggaggctttttcgttatttccaaatacttgagaaattaccttttggtttac 118
|||||||||||||||||||||
||||||||||||||||||||||||||||||||||||||
Sbjct: 3524 tggtggtagggaggctttttc-ttatttccaaatacttgagaaattaccttttggtttac
3466
Query: 119
aaatctatgatcaacttattccattaaatagatacattnnnnnnnttaaaaactgattct 178
|||||||||||||||||||||||||||||||||||||| |||||||||||||||
Sbjct: 3465 aaatctatgatcaacttattccattaaatagatacattaaaaaaattaaaaactgattct
3406
Query: 179
tctgcagagcactggtgtttctttttataaccccttgaaacaagtctctcacctgagcct 238
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct: 3405 tctgcagagcactggtgtttctttttataaccccttgaaacaagtctctcacctgagcct
3346
Query: 239
gtctaaactttcggagggagtttattattgagtctttatctgtgacagtatttggagatt 298
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct: 3345 gtctaaactttcggagggagtttattattgagtctttatctgtgacagtatttggagatt
3286
Query: 299
tagggatttgatacttaggcctttgaattttagaatacaaaaagagaagca 349
|||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct: 3285 tagggatttgatacttaggcctttgaattttagaatacaaaaagagaagca 3235
Les
fichiers de sortie de Blast (BlastN, TblastN, BlastP…) se présentent tous de la
même façon que celui-ci . On trouve :
-
en
entête : la version de Blast
utilisée et l’article correspondant, l’entête du fichier de la séquence soumise
(Query) et la longueur de cette séquence, et la banque de données dans laquelle
Blast doit rechercher les similarités
-
la liste
des séquences montrant le plus de similarité avec la séquence soumise, triées
en fonction de l’expect
-
le
détail des zones de correspondance entre la séquence soumise et chacune des
séquences de la liste précédente (ici le fichier a été tronqué pour ne montrer
que le détail de la meilleure correspondance).
On voit
donc ici apparemment deux localisations différentes sur le même
chromosome : NT009471_17 et NT009471_16. En réalité, il s’agit d’un
artefact dû à la façon dont les contigs sont stockés. En effet, comme il s’agit
de très longues séquences, les contigs sont subdivisés en fragments de 110 000
bp, qui se chevauchent sur 10 000 bp. Ici, la séquence homologue à notre cDNA
dans le génome humain se trouve dans la zone de chevauchement des fragments 16
et 17 du contig NT009471.
Le cDNA
étudié est donc localisé sans ambiguïté sur le fragment 17 (par exemple) du
contig numéro NT009471 du chromosome 12, du nucléotide n°3 584 au nucléotide n°3 235
(c’est-à-dire sur le brin Minus, brin complémentaire de celui dont la séquence
est stockée dans le fichier NT009471_17 de la banque de données du génome
humain).
BlastN contre les ESTs
Première
méthode pour trouver la fonction : on soumet la séquence donnée par
RepeatMasker au programme BlastN pour rechercher les séquences homologues dans
les ESTs. On a le fichier de sortie suivant (extrait) :
BLASTN 2.2.1 [Aug-1-2001]
Reference: Altschul, Stephen F., Thomas L. Madden,
Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997),
"Gapped
BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query=
Homo sapiens cDNA (masked)
(764 letters)
Database:
Expressed Sequence Tags
8,282,804 sequences; 3,678,041,963
total letters
Searching...................................................done
Score E
Sequences
producing significant alignments: (bits)
Value
GB_EST105:BG739739
BG739739 602630444F1 NCI_CGAP_Skn3 Homo sapie... 650 0.0
GB_EST25:AI827881
AI827881 wf04d10.x1 Soares_NFL_T_GBC_S1 Homo s... 599 e-168
GB_EST31:AU151173 AU151173 AU151173 NT2RP2 Homo sapiens
cDNA clo... 591 e-166
GB_EST95:BF983061 BF983061 602309018F1 NIH_MGC_88 Homo
sapiens c... 575 e-161
GB_EST25:AI808020 AI808020 wf53c06.x1 Soares_NFL_T_GBC_S1
Homo s... 573 e-161
GB_EST13:AA931878 AA931878 oo58f06.s1 NCI_CGAP_Lu5 Homo
sapiens ... 480 e-133
GB_EST43:AW157153 AW157153 au92b03.x1 Schneider fetal
brain 0000... 470 e-130
GB_EST47:AW467264 AW467264 he08c09.x1 NCI_CGAP_CML1 Homo
sapiens... 462 e-127
GB_EST22:AI632989 AI632989 tz33b04.x1 NCI_CGAP_Ut2 Homo
sapiens ... 444 e-122
GB_EST99:BG290803 BG290803 602389135F1 NIH_MGC_93 Homo
sapiens c... 440 e-121
GB_EST31:AU146213 AU146213 AU146213 HEMBA1 Homo sapiens
cDNA clo... 404 e-110
GB_EST117:W94441 W94441 ze12b02.r1
Soares_fetal_heart_NbHH19W Ho...
361 5e-97
GB_EST117:W94256 W94256 ze12b02.s1
Soares_fetal_heart_NbHH19W Ho...
353 1e-94
GB_EST31:AU152388 AU152388 AU152388 NT2RP3 Homo sapiens
cDNA clo... 349 2e-93
GB_EST23:AI694590 AI694590 wd88f05.x1 NCI_CGAP_Lu24 Homo
sapiens... 329 2e-87
GB_EST19:AI376246 AI376246 ta55f04.x1
Soares_total_fetus_Nb2HF8_... 315 2e-83
GB_EST91:BF679650 BF679650 602154302F1 NIH_MGC_83 Homo
sapiens c... 303 9e-80
GB_EST25:AI809272 AI809272 wf69h02.x1 Soares_NFL_T_GBC_S1
Homo s... 283 9e-74
GB_EST23:AI680057 AI680057 tw64e07.x1 NCI_CGAP_Ut3 Homo
sapiens ... 283 9e-74
GB_EST18:AI270490 AI270490 qu85a06.x1 NCI_CGAP_Gas4 Homo
sapiens... 283 9e-74
GB_EST2:AA126954 AA126954 zl87g04.s1 Stratagene colon
(#937204) ... 281 3e-73
GB_EST42:AW072665 AW072665 xa41e09.x1 NCI_CGAP_Sar4 Homo
sapiens... 280 1e-72
GB_EST41:AW007290 AW007290 wt54f04.x1 NCI_CGAP_Pan1 Homo
sapiens... 280 1e-72
GB_EST11:AA746226 AA746226 ob22c08.s1 NCI_CGAP_Kid5 Homo
sapiens... 280 1e-72
GB_EST11:AA721330 AA721330 nz73a03.s1 NCI_CGAP_GCB1 Homo
sapiens... 280 1e-72
GB_EST10:AA683509
AA683509 zf34a05.s1 Soares_fetal_heart_NbHH19W... 278 5e-72
GB_EST7:AA447739
AA447739 aa18f04.s1 Soares_NhHMPu_S1 Homo sapie... 278 5e-72
>GB_EST105:BG739739
BG739739 602630444F1 NCI_CGAP_Skn3 Homo sapiens
cDNA clone IMAGE:4775842 5', mRNA
sequence. 5/2001
Length = 764
Score =
650 bits (328), Expect = 0.0
Identities = 342/349 (97%)
Strand = Plus /
Plus
Query: 1
gtaagggtgtactaggggataggatgatgtaagagaatgagaaagatgaccaaaaggttg 60
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct: 1
gtaagggtgtactaggggataggatgatgtaagagaatgagaaagatgaccaaaaggttg 60
Query: 61
gtggtagggaggctttttcgttatttccaaatacttgagaaattaccttttggtttacaa 120
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct: 61
gtggtagggaggctttttcgttatttccaaatacttgagaaattaccttttggtttacaa 120
Query: 121
atctatgatcaacttattccattaaatagatacattnnnnnnnttaaaaactgattcttc 180
|||||||||||||||||||||||||||||||||||| |||||||||||||||||
Sbjct: 121
atctatgatcaacttattccattaaatagatacattaaaaaaattaaaaactgattcttc 180
Query: 181
tgcagagcactggtgtttctttttataaccccttgaaacaagtctctcacctgagcctgt 240
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct: 181 tgcagagcactggtgtttctttttataaccccttgaaacaagtctctcacctgagcctgt
240
Query: 241
ctaaactttcggagggagtttattattgagtctttatctgtgacagtatttggagattta 300
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct: 241 ctaaactttcggagggagtttattattgagtctttatctgtgacagtatttggagattta
300
Query: 301
gggatttgatacttaggcctttgaattttagaatacaaaaagagaagca 349
|||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct: 301
gggatttgatacttaggcctttgaattttagaatacaaaaagagaagca 349
Score = 113 bits
(57), Expect = 2e-22
Identities = 57/57 (100%)
Strand = Plus /
Plus
Query: 708 ctgggagcacacacgaccacgggggccaaagaacggtaaaacatagacgaacacaag
764
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct: 708
ctgggagcacacacgaccacgggggccaaagaacggtaaaacatagacgaacacaag 764
Score = 38.2 bits (19), Expect = 8.5
Identities = 19/19 (100%)
Strand = Plus /
Plus
Query: 665 gagacgccggggggaccat 683
|||||||||||||||||||
Sbjct: 665 gagacgccggggggaccat 683
La
meilleure séquence homologue de notre cDNA dans les ESTs (en terme d’expect)
est l’EST numéro GB_EST105:BG739739. La définition de cette séquence est la
suivante : « 602630444F1 NCI_CGAP_Skn3 Homo sapiens cDNA clone
IMAGE:4775842 5', mRNA sequence ». Cette définition ne nous apporte pas
d’information sur la fonction du cDNA. On compare donc maintenant le cDNA aux
séquences de la base de données GenEmbl.
BlastN contre GenEmbl
Deuxième
méthode pour trouver la fonction : on soumet la séquence donnée par
RepeatMasker au programme BlastN pour rechercher les séquences homologues dans
la banque GenEmbl. On a le fichier de sortie suivant:
BLASTN 2.2.1 [Aug-1-2001]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped
BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query=
Homo sapiens cDNA (masked)
(764 letters)
Database:
GenEMBL
4,352,439 sequences; 11,684,726,444
total letters
Searching...................................................done
Score E
Sequences
producing significant alignments: (bits)
Value
GB_HTG9:AC024896
AC024896 Homo sapiens chromosome 12 clone RP11-... 607 e-170
GB_HTG8:AC023261
AC023261 Homo sapiens chromosome 7 clone RP11-3... 607 e-170
GB_PR6:AK023085
AK023085 Homo sapiens cDNA FLJ13023 fis, clone N... 607 e-170
GB_PR1:AB032470 AB032470 Homo sapiens mRNA for seven
transmembra... 319 5e-84
GB_NEW07:BC005176
BC005176 Homo sapiens, seven transmembrane pro... 266 7e-68
GB_PR8:BC005176 BC005176 Homo sapiens, seven transmembrane prote... 266
7e-68
GB_PR6:AK002031 AK002031 Homo sapiens cDNA FLJ11169 fis, clone P... 262 1e-66
GB_HTG15:AC074091
AC074091 Homo sapiens chromosome 2 clone RP11-... 46 0.12
GB_NEW08:AL512625_3
Continuation (4 of 5) of AL512625 from base ... 44 0.45
GB_NEW08:AL512625_0
AL512625 Homo sapiens chromosome 9 clone RP1... 44 0.45
GB_NEW07:AC068255
AC068255 Homo sapiens chromosome 18 clone RP11... 44 0.45
GB_HTG23:AP001896
AP001896 Homo sapiens chromosome 18 clone RP11... 44 0.45
GB_HTG22:AL590399
AL590399 Homo sapiens chromosome 9 clone RP11-... 44 0.45
GB_HTG22:AL512625
AL512625 Homo sapiens chromosome 9 clone RP11-... 44 0.45
GB_HTG21:AL359312
AL359312 Homo sapiens chromosome 9 clone RP11-... 44 0.45
GB_HTG20:AL163539
AL163539 Homo sapiens chromosome 9 clone RP11-... 44 0.45
GB_HTG17:AC087737
AC087737 Homo sapiens chromosome 15 clone RP11... 44 0.45
GB_HTG13:AC068255
AC068255 Homo sapiens chromosome 18 clone RP11... 44 0.45
GB_HTG10:AC026515
AC026515 Homo sapiens chromosome 15 clone RP11... 44 0.45
GB_HTG10:AC025919
AC025919 Homo sapiens chromosome 15 clone RP11... 44 0.45
GB_PR4:AC024576
AC024576 Homo sapiens chromosome 5 clone CTD-234... 44 0.45
GB_GSS11:AQ767127
AQ767127 HS_2206_B1_B03_MR CIT Approved Human ... 42 1.8
GB_PR1:AC002378
AC002378 Human PAC clone RP3-438O4 from 22q12.1-... 42 1.8
>GB_HTG9:AC024896
AC024896 Homo sapiens chromosome 12 clone RP11-421F16,
WORKING DRAFT SEQUENCE, 8
unordered pieces. 12/2000
Length = 159398
Score =
607 bits (306), Expect = e-170
Identities = 341/351 (97%), Gaps = 3/351 (0%)
Strand = Plus / Minus
Query: 1
gtaagggtgtactaggggat-aggatgat-gtaagagaatgagaaagatgaccaaaaggt 58
||||||||||||||||||||
|||||||| ||||||||||||||||||||||||||||||
Sbjct: 44515 gtaagggtgtactaggggattaggatgattgtaagagaatgagaaagatgaccaaaaggt
44456
Query: 59
tggtggtagggaggctttttcgttatttccaaatacttgagaaattaccttttggtttac 118
|||||||||||||||||||||
||||||||||||||||||||||||||||||||||||||
Sbjct: 44455 tggtggtagggaggctttttc-ttatttccaaatacttgagaaattaccttttggtttac
44397
Query: 119
aaatctatgatcaacttattccattaaatagatacattnnnnnnnttaaaaactgattct 178
|||||||||||||||||||||||||||||||||||||| |||||||||||||||
Sbjct: 44396 aaatctatgatcaacttattccattaaatagatacattaaaaaaattaaaaactgattct
44337
Query: 179
tctgcagagcactggtgtttctttttataaccccttgaaacaagtctctcacctgagcct 238
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct: 44336 tctgcagagcactggtgtttctttttataaccccttgaaacaagtctctcacctgagcct
44277
Query: 239
gtctaaactttcggagggagtttattattgagtctttatctgtgacagtatttggagatt 298
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct: 44276 gtctaaactttcggagggagtttattattgagtctttatctgtgacagtatttggagatt
44217
Query: 299
tagggatttgatacttaggcctttgaattttagaatacaaaaagagaagca 349
|||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct: 44216 tagggatttgatacttaggcctttgaattttagaatacaaaaagagaagca 44166
La
meilleure séquence homologue de notre cDNA dans les GenEmbl (en terme d’expect)
est la séquence numéro GB_HTG9:AC024896. La définition de cette séquence est la suivante : Homo sapiens chromosome 12 clone
RP11-421F16, WORKING DRAFT SEQUENCE, 8 unordered pieces.». Si cette définition nouis
confirme la localisation sur le chromosome 12, elle ne nous apporte pas non
plus d’information sur la fonction du cDNA. On utilise donc la troisième
méthode.
GenScan
Troisième méthode pour trouver la fonction : on soumet
le fragment 17 du contig
numéro NT009471 du
chromosome 12 (voir le résultat du BlastN contre le génome humain) au programme
GenScan. On obtient le fichier de sortie suivant :
GENSCAN
1.0 Date run: 29-Aug-101 Time: 12:17:49
Sequence
Unknown : 110000 bp : 40.04% C+G : Isochore 1 ( 0 - 43 C+G%)
Parameter matrix: HumanIso.smat
Predicted
genes/exons:
Gn.Ex
Type S .Begin ...End .Len Fr Ph I/Ac Do/T CodRg P.... Tscr..
-----
---- - ------ ------ ---- -- -- ---- ---- ----- ----- ------
1.11 PlyA -
1364 1359 6 1.05
1.10 Term -
3989 3727 263
0 2 83 37 279 0.909
17.00
1.09 Intr -
5420 5258 163
2 1 113 95 114 0.999
13.23
1.08 Intr -
9282 9144 139
2 1 29 80 115 0.086
4.35
1.07 Intr -
9858 9546 313
1 1 32 57 144 0.066
0.02
1.06 Intr -
17994 17817 178
0 1 106 95 20 0.627
3.07
1.05 Intr -
22775 22604 172
1 1 41 94 127 0.814
7.62
1.04 Intr -
24229 24109 121
2 1 79 94
82 0.989 6.53
1.03 Intr -
27043 26893 151
1 1 80 76 72 0.441
4.01
1.02 Intr -
30757 30603 155
2 2 53 131 46 0.335
4.27
1.01 Init -
41535 41445 91
0 1 94 86 164 0.185
17.65
1.00 Prom -
44221 44182 40 -7.95
2.00 Prom +
46751 46790 40 -6.65
2.01 Init +
49947 49988 42
2 0 100 85 56 0.480
7.16
2.02 Intr +
53787 53901 115
2 1 62 92 57 0.798
2.60
2.03 Intr +
54714 54814 101
1 2 55 115 144 0.935
12.71
2.04 Term +
55652 55828 177
1 0 17 39 223 0.932
7.00
2.05 PlyA +
57364 57369 6 1.05
3.04 PlyA -
57928 57923 6 1.05
3.03 Term -
64335 63574 762
0 0 83 48 197 0.957
7.63
3.02 Intr -
64820 64391 430
1 1 2 86 196 0.540
3.59
3.01 Init -
67495 67218 278
1 2 47 103 202 0.918
14.10
3.00 Prom -
68367 68328 40 -3.65
4.00 Prom +
69849 69888 40 -6.15
4.01 Sngl +
70303 70959 657
0 0 66 53 249 0.489
15.12
4.02 PlyA +
73443 73448 6 1.05
5.07 PlyA -
73878 73873 6 1.05
5.06 Term -
91568 90897 672
2 0 -175 54
902 0.583 53.26
5.05 Intr -
94682 94591 92
0 2 1 85 109 0.531
0.69
5.04 Intr -
95886 95820 67
0 1 122 91 59 0.552
7.16
5.03 Intr - 101525 101432 94
1 1 38 50 106 0.025
0.85
5.02 Intr - 109229 109105 125
2 2 91 43 90 0.021
3.26
5.01 Intr - 109941 109730 212
1 2 28 82 176 0.337
8.71
Predicted
peptide sequence(s):
>HS12:NT009471_17|GENSCAN_predicted_peptide_1|581_aa
MGFLQLLVVAVLASEHRVAGAAEVFGNSSEGLIEFSVGKFRYFELNRPFPEEAILHDISS
NVTFLIFQIHSQYQNTTVSFSPTLLSNSSETGTASGLVFILRPEQSTCTWYLGTSGIQPV
QNMAILLSYSERDPVPGGCNLEFDLDIDPHITWEYNSFETTIKFAPANLGYARGVDPPPC
DAGTDQDSRWRLQYDVYQYFLPENDLTEEMLLKHLQRMVSVPQVKASALKVVTLTANDKT
SVSFSSLPGQGVIYNVIVWDPFLNTSAAYIPAHTYACSFEAGEGSCASLERVALCWLHGT
TEEPSVPDLQKAEAGTGNGIVCLRHAVGSTAGYPAVHPLSLYSESDSDSCHWKRRWNVLG
SCVVAIWNPLDLHALCWTSAGVPHLVSDFLYSTGNNGGKGNKEVKALMAAESGNLVLCFW
SSVVPSTSTPIRCQTPGVPQLNILTCGVIGSYSVVLAIDSYWSTSLSYITLNVLKRALNK
DFHRAFTNVPFQTNDFIILAVWGMLAVSGITLQIRRERGRPFFPPHPYKLWKQERERRVT
NILDPSYHIPPLRERLYGRLTQIKGLFQKEQPAGERTPLLL
>HS12:NT009471_17|GENSCAN_predicted_peptide_2|144_aa
MADRLTQLQDAVNSLADQFCNAIGVLQQCGPPASFNNIQTAINKDQPANPTEEYAQLFAA
LIARTAKDIDVLIDSLPSEESTAALQAASLYKLEEENHEAATCLEDVVYRGDMLLEKIQS
ALADIAQSQLKTRSGTHSQSLPDS
>HS12:NT009471_17|GENSCAN_predicted_peptide_3|489_aa
MTRGKQNTPKKSATTKQGTEEARRKKRRRTAQRNEGKREEERAKDRKKRGRGKEESRKRA
TGSAHKKKRPDTGRQPTQTKKKSTRRKKGRKNRAIRQDKEIKGIQLGKEEVKLSLFADDM
TVCIENPIVSAQNLLKLISNFSKVSGYKINVQKSQAFLYTDNRQTESQIMSELPFTIASK
RIKYLGIQLTRDVKDLFKKNYKPLLKEIKEDTNKWKNIPCSSVGRINIVKMAILPKNWKK
TTLKFIWNQKGTHISKSILSQKNKAGGITLPDFKLYYKATVTKTAWYWYQNRDINQWNRT
EASEITPHIYNYLIFDKPEKNKQWGKDSLFNKRCWENWLAICRKLKLDPFFTAYTKINSR
WIKDLNVRPKTIKTLEENLDITIQDIGMGKDFMSETPKAMAIKAKIDKWDLIKLKSFCTA
KETTIRVNRQPTEWEKIFATYSSDKGLISRIYSELQQIYKKKPNNPIKKWAKDMNRHFSK
EDIYAAKRH
>HS12:NT009471_17|GENSCAN_predicted_peptide_4|218_aa
MKAEIKVFFETNENKDTTYQNLWDTFQAVCRGKFIALNAHKRKQERSKIDTLTSQLKELE
KQEQTHSKASRRQEITKIRAELKEIETQKTLQKINESRSWFFEKINKIDRPLASLIKKKR
EKNQTDAIKNDKGDITTDPTEIQTTIREYCKYLCANKLENLEEMDKFLHTYTLPRLNQEE
VESLNRPITGSEIEAIINSLPIKKRPHGIFMECKSVYR
>HS12:NT009471_17|GENSCAN_predicted_peptide_5|420_aa
XSRANRLLNGDNLWIDKLPKERTKLSVGKLNNLVQEFQIFLENLKDDDAVFPETAQQDFQ
LSSGSPPEMVQMISQATASQRTSAPEISSILSEQPEKDDTPSHTQAQCCLNFGTNDKSHM
IISIVAEKAFDKIQHPFMLKTLNETHRVSQAESSLGLLLFQVLDLIGDRQHAPYLSRILI
QTTEVLTVTLQGCLEDKKEKKRKERGKEGWKEKERNIKNERKKERGGKGGREEKEEGRSR
RRKKKKEEEEEEEEKEEAAEAAAGGGGGRRKQKKKKEEGEGEEEEERRIRRGRRRRRRKE
EEEEGRRRGEGRRRGRGRRRRRRKRKKKRRRRKKGEEEGGGGGRRKKKEKEEERRRRKKK
EEKEEKEKEERRRRGGRGGGTQEEEGEEEEERGGGGEGKKIYKFTFILDFNLCIFVLKSF
Le fichier de sortie de GenScan comprend tout d’abord
la liste des gènes et des exons prédits, avec les caractéristiques suivantes :
Gn.Ex : numéro
du gène, numéro de l’exon
Type : Prom
= promoteur
Init = exon initial
Intr = exon interne
Term = exon terminal
PlyA
= site de polyadénylation
S : Brin d’ADN (+ =
brin donné en entrée, - = brin complémentaire)
Begin : début
de l’exon, du promoteur ou du site de polyadénylation (numéroté
sur le brin donné en entrée)
End : fin de l’exon, du
promoteur ou du site de polyadénylation (numéroté sur
le brin
donné en entrée)
Len : longueur de l’exon,
du promoteur ou du site de polyadénylation (bp)
Fr : cadre de lecture (un
codon se terminant en x est dans le cadre f = x mod
3)
Ph : net phase of exon (longueur mod 3)
I/Ac : score du signal
d’initiation ou du site accepteur d’épissage (x 10)
Do/T : score du site donneur
d’épissage ou du signal de terminaison (x 10)
CodRg : score de la region
codante (x 10)
P : probabilité de
l’exon
Tscr : score total de l’exon
(dépend de la longueur, de I/Ac, de Do/T et de
CodRg)
Le
fichier présente ensuite pour chaque gène détecté la séquence en acides aminés
de la protéine correspondante.
La séquence que nous étudions a été localisée sur le
fragment de contig numéro NT009471_17 du chromosome 2, du nucléotide
n°3 584 au nucléotide n°3 235. Cette séquence serait donc issue du gène prédit
numéro 1. Il faut rechercher les homologues de cette protéine prédite dans les
banques protéiques
BlastP contre les banques protéiques
On soumet donc au programme BlastP la séquence de la
protéine prédite numéro 1, en précisant comme banque de séquences l’ensemble
des banques protéiques. On obtient le fichier suivant (extrait) :
BLASTP 2.2.1 [Aug-1-2001]
Reference: Altschul, Stephen F., Thomas L. Madden,
Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997),
"Gapped
BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query=
HS12:NT009471_17|GENSCAN_predicted_peptide_1|581_aa
(581 letters)
Database:
SwissProt + SPTrEMBL
675,459 sequences; 213,887,202
total letters
Searching..................................................done
Score E
Sequences
producing significant alignments: (bits)
Value
SPT:Q9NS93
Q9ns93 SEVEN TRANSMEMBRANE PROTEIN TM7SF3. 6/2001 838 0.0
SPT:Q9NUS4
Q9nus4 CDNA FLJ11169 FIS, CLONE PLACE1007282. 10/2000
836 0.0
SPT:Q9CRG1 Q9crg1 2010003B14RIK PROTEIN (FRAGMENT).
6/2001 415 e-115
SPT:Q86204
Q86204 VP8 AND VP85 (FRAGMENT). 6/2001 42 0.018
SPT:Q9Q2P6
Q9q2p6 VP4. 6/2001 40 0.052
SPT:Q9WN88
Q9wn88 VP4 PROTEIN (FRAGMENT). 6/2001 40
0.068
SPT:O90365
O90365 OUTER CAPSID PROTEIN VP4. 6/2001 40
0.068
SPT:Q86201 Q86201 OUTER CAPSID PROTEIN. 6/2001 40 0.068
SW:VP4_ROTHT P11200 OUTER CAPSID PROTEIN VP4
(HEMAGGLUTININ) (OU... 40 0.068
SPT:O90315 O90315 OUTER CAPSID PROTEIN VP4. 6/2001 39 0.089
SPT:Q82119 Q82119 OUTER CAPSID PROTEIN VP4. 6/2001 39 0.089
SPTNEW:CAC43311 Cac43311 CAPSID PROTEIN (FRAGMENT).
8/2001 39 0.12
SPT:Q9IV24 Q9iv24 OUTER CAPSID PROTEIN 4. 6/2001 39 0.12
SPT:Q9WN85 Q9wn85 VP4 PROTEIN (FRAGMENT). 6/2001 39 0.12
SPT:Q86223 Q86223 OUTER CAPSID PROTEIN. 6/2001 39 0.12
SPT:Q86222 Q86222 OUTER CAPSID PROTEIN. 6/2001 39 0.12
SPT:Q86221 Q86221 OUTER CAPSID PROTEIN. 6/2001 39
0.12
SPT:Q86206
Q86206 VP8 AND VP85 (FRAGMENT). 6/2001 39
0.12
SPT:Q86205
Q86205 VP8 AND VP85 (FRAGMENT). 6/2001 39
0.12
SPT:Q86203
Q86203 VP8 AND VP85 (FRAGMENT). 6/2001 39 0.12
SPT:Q67524 Q67524 VP4 PROTEIN (FRAGMENT). 6/2001 39 0.12
SW:VP4_ROTHM P11197 OUTER CAPSID PROTEIN VP4
(HEMAGGLUTININ) (OU... 39 0.12
SPT:Q9WN84 Q9wn84 VP4 PROTEIN (FRAGMENT). 6/2001 39 0.15
SPT:Q9WN86 Q9wn86 VP4 PROTEIN (FRAGMENT). 6/2001 39 0.15
SPT:Q86200 Q86200 CAPSID PROTEIN. 6/2001 39 0.15
SW:VP4_ROTH1 P11198 OUTER CAPSID PROTEIN VP4
(HEMAGGLUTININ) (OU... 39 0.15
SPT:O90314 O90314 OUTER CAPSID PROTEIN VP4. 6/2001 38 0.20
SW:VP4_ROTHN P11199 OUTER CAPSID PROTEIN VP4
(HEMAGGLUTININ) (OU... 38 0.26
SPT:Q9W9H4 Q9w9h4 VP4 PROTEIN (FRAGMENT). 9/2001 37 0.58
SPT:Q9WN87 Q9wn87 VP4 PROTEIN (FRAGMENT). 6/2001 37 0.58
SPT:Q67526 Q67526 VP4 PROTEIN (FRAGMENT). 6/2001 37 0.58
SPT:Q90100 Q90100 VP4 PROTEIN (FRAGMENT). 6/2001 36 0.76
>SPT:Q9NS93 Q9ns93 SEVEN TRANSMEMBRANE PROTEIN TM7SF3.
6/2001
Length = 570
Score =
838 bits (2166), Expect = 0.0
Identities = 459/593 (77%), Positives = 476/593 (79%),
Gaps = 35/593 (5%)
Query: 1
MGFLQLLVVAVLASEHRVAGAAEVFGNSSEGLIEFSVGKFRYFELNRPFPEEAILHDISS 60
MGFLQLLVVAVLASEHRVAGAAEVFGNSSEGLIEFSVGKFRYFELNRPFPEEAILHDISS
Sbjct: 1
MGFLQLLVVAVLASEHRVAGAAEVFGNSSEGLIEFSVGKFRYFELNRPFPEEAILHDISS 60
Query: 61
NVTFLIFQIHSQYQNTTVSFSPTLLSNSSETGTASGLVFILRPEQSTCTWYLGTSGIQPV 120
NVTFLIFQIHSQYQNTTVSFSPTLLSNSSETGTASGLVFILRPEQSTCTWYLGTSGIQPV
Sbjct: 61
NVTFLIFQIHSQYQNTTVSFSPTLLSNSSETGTASGLVFILRPEQSTCTWYLGTSGIQPV 120
Query: 121
QNMAILLSYSERDPVPGGCNLEFDLDIDPHITWEYNSFETTIKFAPANLGYARGVDPPPC 180
QNMAILLSYSERDPVPGGCNLEFDLDIDP+I
EYN FETTIKFAPANLGYARGVDPPPC
Sbjct: 121 QNMAILLSYSERDPVPGGCNLEFDLDIDPNIYLEYNFFETTIKFAPANLGYARGVDPPPC
180
Query: 181
DAGTDQDSRWRLQYDVYQYFLPENDLTEEMLLKHLQRMVSVPQVKASALKVVTLTANDKT 240
DAGTDQDSRWRLQYDVYQYFLPENDLTEEMLLKHLQRMVSVPQVKASALKVVTLTANDKT
Sbjct: 181 DAGTDQDSRWRLQYDVYQYFLPENDLTEEMLLKHLQRMVSVPQVKASALKVVTLTANDKT
240
Query: 241
SVSFSSLPGQGVIYNVIVWDPFLNTSAAYIPAHTYACSFEAGEGSCASLERVA------L 294
SVSFSSLPGQGVIYNVIVWDPFLNTSAAYIPAHTYACSFEAGEGSCASL RV+ L
Sbjct: 241
SVSFSSLPGQGVIYNVIVWDPFLNTSAAYIPAHTYACSFEAGEGSCASLGRVSSKVFFTL 300
Query: 295
CWLHGTTEEPSVPDLQKAEA-GTGNGIVCLRHAVGSTAGYPAVHPLSLYSESDSDSCHWK 353
L
G K E G
I+ + T
P + ++L + + S
Sbjct: 301
FALLGFFICFFGHRFWKTELFFIGFIIMGFFFYILITRLTPIKYDVNLILTAVTGS---- 356
Query: 354 RRWNVLGSCVVAIWNPLDLHALCWTSAGVPHLVSDFLYSTXXXXXXXXXEVKALMAAESG
413
V
G +VA+W + ++C G LV
FL S+ G
Sbjct: 357
----VGGMFLVAVWWRFGILSICMLCVG---LVLGFLISS------------VTFFTPLG 397
Query: 414
NLVLCFWSSVVPSTSTPIRCQTPGVPQ-----LNILTCGVIGSYSVVLAIDSYWSTSLSY 468
NL
+ V T + I P V LNILTCGVIGSYSVVLAIDSYWSTSLSY
Sbjct: 398
NLKIFHDDGVFWVTFSCIAILIPVVFMGCLRILNILTCGVIGSYSVVLAIDSYWSTSLSY 457
Query: 469
ITLNVLKRALNKDFHRAFTNVPFQTNDFIILAVWGMLAVSGITLQIRRERGRPFFPPHPY 528
ITLNVLKRALNKDFHRAFTNVPFQTNDFIILAVWGMLAVSGITLQIRRERGRPFFPPHPY
Sbjct: 458
ITLNVLKRALNKDFHRAFTNVPFQTNDFIILAVWGMLAVSGITLQIRRERGRPFFPPHPY 517
Query: 529
KLWKQERERRVTNILDPSYHIPPLRERLYGRLTQIKGLFQKEQPAGERTPLLL 581
KLWKQERERRVTNILDPSYHIPPLRERLYGRLTQIKGLFQKEQPAGERTPLLL
Sbjct: 518
KLWKQERERRVTNILDPSYHIPPLRERLYGRLTQIKGLFQKEQPAGERTPLLL 570
La
meilleure correspondance de la protéine prédite par GenScan dans les banques
protéiques est donc la protéine SPT:Q9NS93. En interrogeant directement
la banque protéique avec ce numéro, on obtient la fiche suivante :
ID Q9NS93
PRELIMINARY; PRT; 570 AA.
AC Q9NS93;
DT 01-OCT-2000 (TrEMBLrel. 15, Created)
DT 01-OCT-2000 (TrEMBLrel. 15, Last sequence
update)
DT 01-JUN-2001 (TrEMBLrel. 17, Last annotation
update)
DE SEVEN TRANSMEMBRANE PROTEIN
TM7SF3.
GN TM7SF3.
OS Homo sapiens (Human).
OC Eukaryota; Metazoa; Chordata; Craniata;
Vertebrata; Euteleostomi;
OC Mammalia; Eutheria; Primates; Catarrhini;
Hominidae; Homo.
OX NCBI_TaxID=9606;
RN [1]
RP SEQUENCE FROM N.A.
RX MEDLINE=20291015; PubMed=10828615;
RA Akashi H., Han H.-J., Iizaka M., Nakajima
Y., Furukawa Y., Sugano S.,
RA Imai K., Nakamura Y.;
RT "Isolation and characterization of a
novel gene encoding a putative
RT seven-span transmembrane protein,
TM7SF3.";
RL Cytogenet. Cell Genet. 88:305-309(2000).
RN [2]
RP SEQUENCE FROM N.A.
RC TISSUE=CHORIOCARCINOMA;
RA Strausberg
R.;
RL Submitted (MAR-2001) to the
EMBL/GenBank/DDBJ databases.
DR EMBL;
AB032470; BAA92856.1; -.
DR EMBL; BC005176; AAH05176.1; -.
KW Transmembrane.
SQ SEQUENCE
570 AA; 64166 MW; CC296D7C22AD894C CRC64;
Q9NS93
Length: 570 August 29, 19101
13:39 Type: P Check: 5683 ..
1
MGFLQLLVVA VLASEHRVAG AAEVFGNSSE GLIEFSVGKF RYFELNRPFP
51
EEAILHDISS NVTFLIFQIH SQYQNTTVSF SPTLLSNSSE TGTASGLVFI
101
LRPEQSTCTW YLGTSGIQPV QNMAILLSYS ERDPVPGGCN LEFDLDIDPN
151
IYLEYNFFET TIKFAPANLG YARGVDPPPC DAGTDQDSRW RLQYDVYQYF
201
LPENDLTEEM LLKHLQRMVS VPQVKASALK VVTLTANDKT SVSFSSLPGQ
251
GVIYNVIVWD PFLNTSAAYI PAHTYACSFE AGEGSCASLG RVSSKVFFTL
301
FALLGFFICF FGHRFWKTEL FFIGFIIMGF FFYILITRLT PIKYDVNLIL
351
TAVTGSVGGM FLVAVWWRFG ILSICMLCVG LVLGFLISSV TFFTPLGNLK
401
IFHDDGVFWV TFSCIAILIP VVFMGCLRIL NILTCGVIGS YSVVLAIDSY
451
WSTSLSYITL NVLKRALNKD FHRAFTNVPF QTNDFIILAV WGMLAVSGIT
501
LQIRRERGRP FFPPHPYKLW KQERERRVTN ILDPSYHIPP LRERLYGRLT
551 QIKGLFQKEQ PAGERTPLLL
La
définition de cette protéine est « SEVEN TRANSMEMBRANE PROTEIN
TM7SF3 ».
On connaît donc la fonction du gène dont provient notre cDNA, à condition que
la prédiction de GenScan soit juste. Il reste à vérifier si cette protéine a
été observée biologiquement, c’est-à-dire s’il existe des ESTs lui
correspondant.
TblastN contre les ESTs
On soumet donc au programme TBlastN la séquence de la
protéine SPT:Q9NS93, en précisant comme banque de séquences celle des ESTs. On
obtient le fichier suivant (extrait) :
TBLASTN 2.2.1 [Aug-1-2001]
Reference: Altschul, Stephen F., Thomas L. Madden,
Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997),
"Gapped
BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query=
SPT:Q9NS93
(570 letters)
Database:
Expressed Sequence Tags
8,282,804 sequences; 3,678,041,963
total letters
Searching..................................................done
Score E
Sequences producing significant alignments: (bits) Value
GB_EST101:BG403521 BG403521 602419218F1 NIH_MGC_93 Homo
sapiens ... 474 e-132
GB_EST30:AU120021 AU120021 AU120021 HEMBA1 Homo sapiens
cDNA clo... 467 e-130
GB_EST108:BI019759 BI019759 IL3-MT0267-110101-442-H09
MT0267 Hom... 451 e-126
GB_EST29:AL550205 AL550205 AL550205 LTI_NFL006_PL2 Homo
sapiens ... 438 e-121
GB_EST96:BG034578 BG034578 602302709F1 NIH_MGC_87 Homo
sapiens c... 438 e-121
GB_EST31:AU135873 AU135873 AU135873 PLACE1 Homo sapiens
cDNA clo... 355 e-121
GB_EST97:BG169037 BG169037 602320444F1 NIH_MGC_89 Homo
sapiens c... 431 e-119
GB_EST31:AU137800 AU137800 AU137800 PLACE1 Homo sapiens
cDNA clo... 424 e-118
GB_EST105:BG722354 BG722354 602693546F1 NIH_MGC_97 Homo
sapiens ... 417 e-117
GB_EST101:BG433331 BG433331 602496716F1 NIH_MGC_75 Homo
sapiens ... 384 e-110
GB_EST95:BF982844 BF982844 602305604F1 NIH_MGC_88 Homo
sapiens c... 397 e-109
GB_EST80:BE875994
BE875994 601486472F1 NIH_MGC_69 Homo sapiens c... 210 e-108
GB_EST108:BI018972 BI018972 IL3-MT0267-050101-437-D05
MT0267 Hom... 391 e-107
GB_EST108:BI019754 BI019754 IL3-MT0267-110101-442-C01
MT0267 Hom... 382 e-105
GB_EST94:BF905903 BF905903 IL3-MT0267-271200-412-F02
MT0267 Homo... 382 e-105
GB_EST99:BG257906 BG257906 602379542F1 NIH_MGC_92 Homo
sapiens c... 365 e-104
GB_EST82:BF034397 BF034397 601455143F1 NIH_MGC_66 Homo
sapiens c... 379 e-104
GB_EST99:BG284187 BG284187 602408236F1 NIH_MGC_91 Homo
sapiens c... 272 e-104
GB_EST80:BE893899
BE893899 601436266F1 NIH_MGC_72 Homo sapiens c... 377 e-103
GB_EST82:BF036852
BF036852 601460327F1 NIH_MGC_66 Homo sapiens c... 377 e-103
GB_EST101:BG403144
BG403144 602418971F1 NIH_MGC_93 Homo sapiens ... 376 e-103
GB_EST99:BG291822
BG291822 602386048F1 NIH_MGC_93 Homo sapiens c... 369 e-101
GB_EST31:AU131888 AU131888 AU131888 NT2RP3 Homo sapiens
cDNA clo... 347 2e-96
GB_EST29:AL550306 AL550306 AL550306 LTI_NFL006_PL2 Homo
sapiens ... 353 2e-96
GB_EST80:BE871129
BE871129 601447386F1 NIH_MGC_65 Homo sapiens c... 280 4e-96
GB_EST31:AU130432 AU130432 AU130432 NT2RP3 Homo sapiens
cDNA clo... 347 3e-94
GB_EST104:BG701006 BG701006 602682187F1 NIH_MGC_95 Homo
sapiens ... 329 7e-89
GB_EST85:BF241654 BF241654 601878995F1 NIH_MGC_55 Homo
sapiens c... 278 8e-87
GB_EST77:BE665527
BE665527 154603 MARC 4BOV Bos taurus cDNA 5', ... 304 2e-81
GB_EST94:BF906228 BF906228 IL3-MT0267-291200-427-H08
MT0267 Homo... 300 2e-80
GB_EST73:BE372157
BE372157 601223770F1 NCI_CGAP_Mam1 Mus musculu... 283 5e-75
GB_EST20:AI464867 AI464867 mz92e09.y1 Soares mouse lymph
node Nb... 280 5e-74
GB_EST108:BI021326 BI021326 IL3-MT0267-160101-449-C04
MT0267 Hom... 279 6e-74
GB_EST73:BE311979
BE311979 601154559F1 NIH_MGC_19 Homo sapiens c... 278 2e-73
GB_EST89:BF568465
BF568465 602184471F1 NIH_MGC_42 Homo sapiens c... 276 7e-73
GB_EST108:BI019341 BI019341 IL3-MT0267-080101-439-A05
MT0267 Hom... 271 2e-71
GB_EST72:BE280022
BE280022 601158480F1 NIH_MGC_21 Homo sapiens c... 270 5e-71
GB_EST15:AI047728 AI047728 uh82e08.r1 Soares mouse
urogenital ri... 268 2e-70
>GB_EST101:BG403521 BG403521 602419218F1 NIH_MGC_93
Homo sapiens
cDNA clone IMAGE:4526165 5', mRNA sequence. 3/2001
Length = 1052
Score =
474 bits (1219), Expect = e-132
Identities = 232/257 (90%), Positives = 234/257 (90%)
Frame = +3
Query: 91
TGTASGLVFILRPEQSTCTWYLGTSGIQPVQNMAILLSYSERDPVPGGCNLEFDLDIDPN 150
TGTASGLVFILRPEQSTCTWYLGTSGIQPVQNMAILLSYSERDPVPGGCNLEFDLDIDPN
Sbjct: 3 TGTASGLVFILRPEQSTCTWYLGTSGIQPVQNMAILLSYSERDPVPGGCNLEFDLDIDPN
182
Query: 151
IYLEYNFFETTIKFAPANLGYARGVDPPPCDAGTDQDSRWRLQYDVYQYFLPENDLTEEM 210
IYLEYNFFETTIKFAPANLGYARGVDPPPCDAGTDQDSRWRLQYDVYQYFLPENDLTEEM
Sbjct: 183 IYLEYNFFETTIKFAPANLGYARGVDPPPCDAGTDQDSRWRLQYDVYQYFLPENDLTEEM
362
Query: 211
LLKHLQRMVSVPQVKASALKVVTLTANDKTSVSFSSLPGQGVIYNVIVWDPFLNTSAAYI 270
LLKHLQRMVSVPQVKASALKVVTLTANDKTSVSFSSLPGQGVIYNVIVWDPFLNTSAAYI
Sbjct: 363
LLKHLQRMVSVPQVKASALKVVTLTANDKTSVSFSSLPGQGVIYNVIVWDPFLNTSAAYI 542
Query: 271
PAHTYACSFEAGEGSCASLGRVSSKVFFTLFALLGFFICFFGHRFWKTEXXXXXXXXXXX 330
PAHTYACSFEAGEGSCASLGRVSSKVFFTL ALLGFF+CFFGHRFWKTE
Sbjct: 543
PAHTYACSFEAGEGSCASLGRVSSKVFFTLIALLGFFMCFFGHRFWKTELFFIGLSSWDS 722
Query: 331 XXXXXXTRLTPIKYDVN 347
PIK+D N
Sbjct: 723 SFIYWLQD*HPIKFDEN 773
On obtient bien des ESTs décrits dans la banque de données
susceptibles de correspondre à la protéine SPT:Q9NS93.